




Nat Chem Biol:针剂转化为口服药片的研究

从 8448 个环肽中,成功打造出一款可口服的凝血酶抑制剂
许多亟需攻克的疾病靶点蛋白,至今缺乏能够口服的药物。有些蛋白表面平坦光滑,小分子难以找到嵌入的口袋;另一些虽存在结合位点,却难以实现高选择性,容易误伤同家族的其他蛋白。过去,这类难题常依赖抗体等大分子生物药——但它们几乎只能通过注射给药。
介于小分子与生物药之间的环肽,原本被视为极具潜力的中间体:尺寸足够小,同时能像大分子一样精准锁定棘手靶点。自然界已经提供了例证——环孢素 A、去氨加压素以及某种生长抑素类似物,都是天然存在且可口服的肽类药物。然而,它们属于少数幸运者。绝大多数天然或人工设计的肽,要么分子偏大、表面亲水性强,要么无法耐受消化道中的蛋白酶,最终只能制成注射剂。

这项来自洛桑联邦理工学院(EPFL)Christian Heinis 团队、发表于《Nature Chemical Biology》的研究,旨在解答一个长期悬而未决的问题:是否能够从零开始,针对特定靶点,设计出既能精准结合、又可口服吸收的小环肽。
三道关卡,相互制约
口服肽类药物之所以困难,在于它必须同时突破三道彼此牵制的障碍。
第一道关卡是消化道内的蛋白酶。胃中的胃蛋白酶、肠道中的胰酶,其天然功能就是将蛋白质和肽链切割成氨基酸。第二道关卡是肠壁:分子必须穿越肠上皮细胞的脂质膜才能被吸收,而肽通常体积大、表面极性基团过多,如同一团亲水线球,难以穿过油性膜层。第三道关卡是肝脏:被吸收进入门静脉的分子,第一站便是肝脏,那里充满了代谢酶,会为分子添加各种修饰、加速其清除,即所谓首过代谢。
研究人员早已总结出可口服肽的典型特征:分子量低于 700 道尔顿、极性表面积小于 200 平方埃、氢键供体不超过五个。但这并非绝对法则——环孢素 A 分子量高达 1,203 道尔顿,却凭借大量 N-甲基化以及可蜷缩隐藏极性表面的构象,仍能实现口服。
真正的难点不在于绘制理想画像,而在于制造出既符合画像特征、又能恰好结合特定靶点的肽。要找到这样的分子,必须从海量候选物中筛选。过去常用噬菌体展示或 mRNA 展示构建大库,但这类生物展示库的化学多样性有限,大多仅使用天然氨基酸,难以得到既短小又高亲和力的结合肽。Heinis 团队此前曾用噬菌体展示开发抗消化道蛋白酶的环肽,即便最小的也有九个氨基酸、分子量 1,134 道尔顿,体积过大且亲水性强,在小鼠体内的口服生物利用度仅有 0.2%。
从二硫键转向硫醚键
Heinis 团队此前的策略,是在微孔板中利用声波分液技术,以纳摩尔级微量规模逐一合成环肽,粗产物不经纯化直接用于筛选——他们曾以此方式造出两万个环肽,并发现了纳摩尔级的结合分子。然而,这批环肽采用二硫键环化,而二硫键存在致命缺陷:在还原环境中易被打开。
这恰恰是口服途径的拦路虎。分子穿越细胞时,细胞内是还原性环境,二硫键会被还原、环状结构解体,肽随之散架。因此,这套高效的合成筛选方法一直无法用于口服肽的研发。这项工作的关键一步,便是将环化所用的化学键从二硫键更换为硫醚键。
硫醚环化还有一个对建库极为有利的优点:反应干净且高效。两端带有巯基的线性肽,与一个含有两个反应位点的连接子混合后,几乎可以定量完成环化。正因为反应副产物少,粗产物中杂质含量低,才敢不纯化直接用于筛选——这是实现规模化、构建大文库的前提。
不过,硫醚键的弱点也需提前摸清:硫原子容易被氧化,而氧化主要来自肝脏中 CYP450 家族酶系,即首过代谢这一关。团队首先合成了十个结构各异的双硫醚环肽 1–10,将其置入大鼠肝微粒体(从大鼠肝细胞中分离、富含代谢酶的组分,常用于预测肝脏代谢清除速率)中,观察其耐受性。结果差异显著:最不稳定的肽 1 半衰期仅约 11 分钟,最稳定的肽 2 则长达 133 分钟;被氧化的肽会多出 16 或 32 的质量,对应添加一个或两个氧原子。以两种已上市口服药维拉帕米和地尔硫卓作为基准,十个环肽中有九个与其相当或更稳定,其中最稳定的四种清除率甚至慢四倍以上。
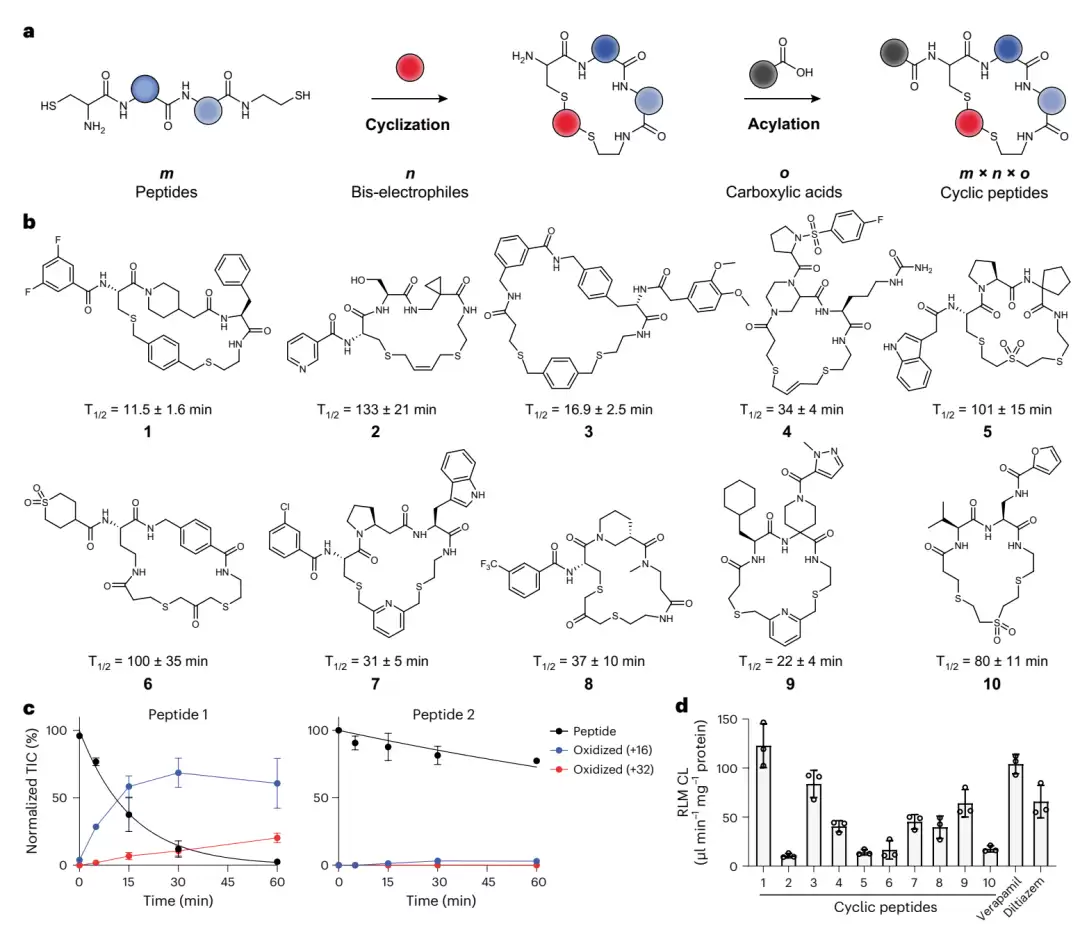 双硫醚环肽的合成思路与代谢稳定性。图 a 展示了整体建库策略:两端带巯基的线性肽先通过双亲电连接子环化,随后在外围氨基上连接一个羧酸,m 种肽 × n 种连接子 × o 种羧酸,组合出规模庞大的环肽库。图 b 列出了十个随机环肽 1–10 的结构及其在大鼠肝微粒体中的半衰期,可直观看出稳定性差异巨大。图 d 将十个肽的清除率与两种口服药进行对比,显示哪些肽比药物更稳定。
双硫醚环肽的合成思路与代谢稳定性。图 a 展示了整体建库策略:两端带巯基的线性肽先通过双亲电连接子环化,随后在外围氨基上连接一个羧酸,m 种肽 × n 种连接子 × o 种羧酸,组合出规模庞大的环肽库。图 b 列出了十个随机环肽 1–10 的结构及其在大鼠肝微粒体中的半衰期,可直观看出稳定性差异巨大。图 d 将十个肽的清除率与两种口服药进行对比,显示哪些肽比药物更稳定。
双硫醚环肽的合成思路与代谢稳定性。图 a 展示了整体建库策略:两端带巯基的线性肽先通过双亲电连接子环化,随后在外围氨基上连接一个羧酸,m 种肽 × n 种连接子 × o 种羧酸,组合出规模庞大的环肽库。图 b 列出了十个随机环肽 1–10 的结构及其在大鼠肝微粒体中的半衰期,可直观看出稳定性差异巨大。图 d 将十个肽的清除率与两种口服药进行对比,显示哪些肽比药物更稳定。
不纯化,直接筛选
要将这套思路转化为可规模化合成的方法,核心在于将两步反应串联起来,且中间不进行纯化:先完成硫醚环化,紧接着在环肽外围的氨基上连接羧酸(酰化)。两步反应只需按顺序向微孔板中加入试剂即可。
这两步反应过去虽各自被使用过,但从未被串联在一起。团队先用八个两端带巯基、且含一个氨基的随机肽(P1–P8)进行探索。这些肽在 96 孔板中通过半胱胺树脂合成,依靠乙醚沉淀即可获得不错的纯度,八个肽的平均纯度达到 93%,全程无需色谱纯化。
接着测试环化效率:将八个肽分别与四个连接子(L1–L4)两两组合,共 32 个反应。为确保分子内成环优于分子间副反应,反应在低浓度(1 毫摩尔)、大体积(1 毫升)下进行,连接子用量为两倍。32 个反应的环化效率平均为 85%,最主要的副产物是二硫键环化的肽,在大多数反应中占比不到 1%,最高也只有 15%。随后测试酰化:将环化完成的肽与八个羧酸(A1–A8)组合,在 384 孔板中以 500 皮摩尔的微量、50 纳升的反应体积进行,室温反应 8 小时,结果几乎实现定量酰化。
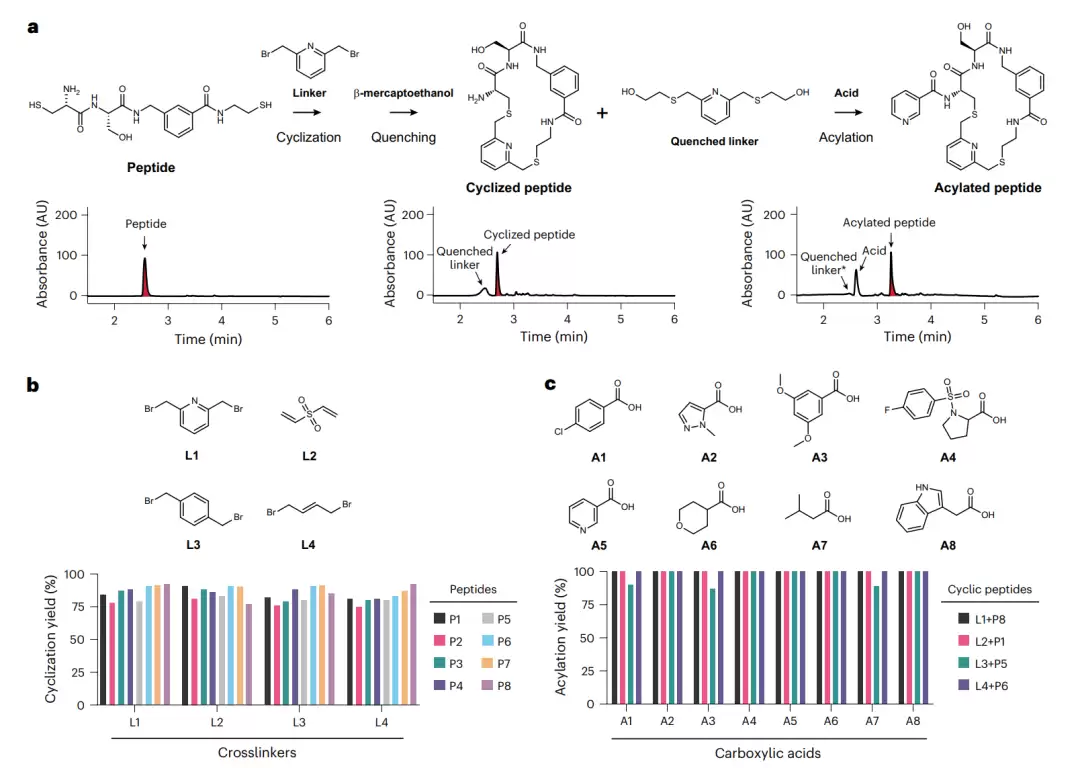 环化与酰化两步反应的效率验证。图 a 以示例肽展示完整流程:线性肽先环化,用 β-巯基乙醇淬灭多余连接子,再进行酰化,三张色谱图分别对应三步后的产物。图 b 展示了八个肽与四个连接子的环化产率,图 c 展示了酰化产率,可见两步反应对不同序列和试剂均具有广泛的适应性。
环化与酰化两步反应的效率验证。图 a 以示例肽展示完整流程:线性肽先环化,用 β-巯基乙醇淬灭多余连接子,再进行酰化,三张色谱图分别对应三步后的产物。图 b 展示了八个肽与四个连接子的环化产率,图 c 展示了酰化产率,可见两步反应对不同序列和试剂均具有广泛的适应性。
环化与酰化两步反应的效率验证。图 a 以示例肽展示完整流程:线性肽先环化,用 β-巯基乙醇淬灭多余连接子,再进行酰化,三张色谱图分别对应三步后的产物。图 b 展示了八个肽与四个连接子的环化产率,图 c 展示了酰化产率,可见两步反应对不同序列和试剂均具有广泛的适应性。
八千多个环肽,瞄准凝血酶
有了合成方法,团队选择凝血酶作为模型靶点进行验证。
不带电的口服凝血酶抑制剂一直难以开发。Heinis 团队此前制备的凝血酶环肽,要么带有电荷(无法穿透细胞膜),要么采用二硫键环化(不耐还原)。此次他们专门设计了一个文库:不含任何电荷,采用硫醚键环化。每个肽由六个构件组成——两个随机氨基酸、两个固定巯基构件(如半胱氨酸)、一个随机连接子、一个随机外围羧酸。十个备选羧酸中包括 A11,它本身能够微弱地结合凝血酶的 S1 口袋。
他们首先合成了 384 个两端带巯基的短肽(四种格式理论上可组出 400 个,实际获得 384 个),平均产量为 4.4 微摩尔,不经纯化;随后使用两个连接子进行环化,十个羧酸(外加不加酸的对照)进行酰化,最终得到 8,448 个环肽,全部直接保留在合成板中待筛选(384 × 2 × 11 = 8,448)。这批环肽的物化性质分析显示,绝大多数超出了“五规则”的范围——虽然分子量不大,却与经典小分子有明显差异。
筛选直接在合成板中进行:向反应孔中加入凝血酶以及一种遇酶切割会发荧光的底物,将肽稀释至 10 微摩尔,检测残余的凝血酶活性。在 8,448 个反应中,有 73 个(0.9%)将凝血酶活性抑制了一半以上。将结果绘制成热图,可观察到非常整齐的规律:所有最活跃的肽,外围连接的均是 A11 或 A12 这两个羧酸。对最强的 20 个命中肽重新合成并复测,活性几乎不变,说明这套合成与筛选方法非常稳健。
对这 20 个肽进行构效分析,归纳出六组结构相似的肽(11–30)。重新合成纯化后测定抑制常数(Ki),均落在纳摩尔级别,其中环肽 11 表现最佳,对人 α-凝血酶的 Ki 为 82 ± 10 纳摩尔。最强的第一组(11–16)共享一个 Cys-Phe 基序,半胱氨酸的氨基上连接着氯噻吩。
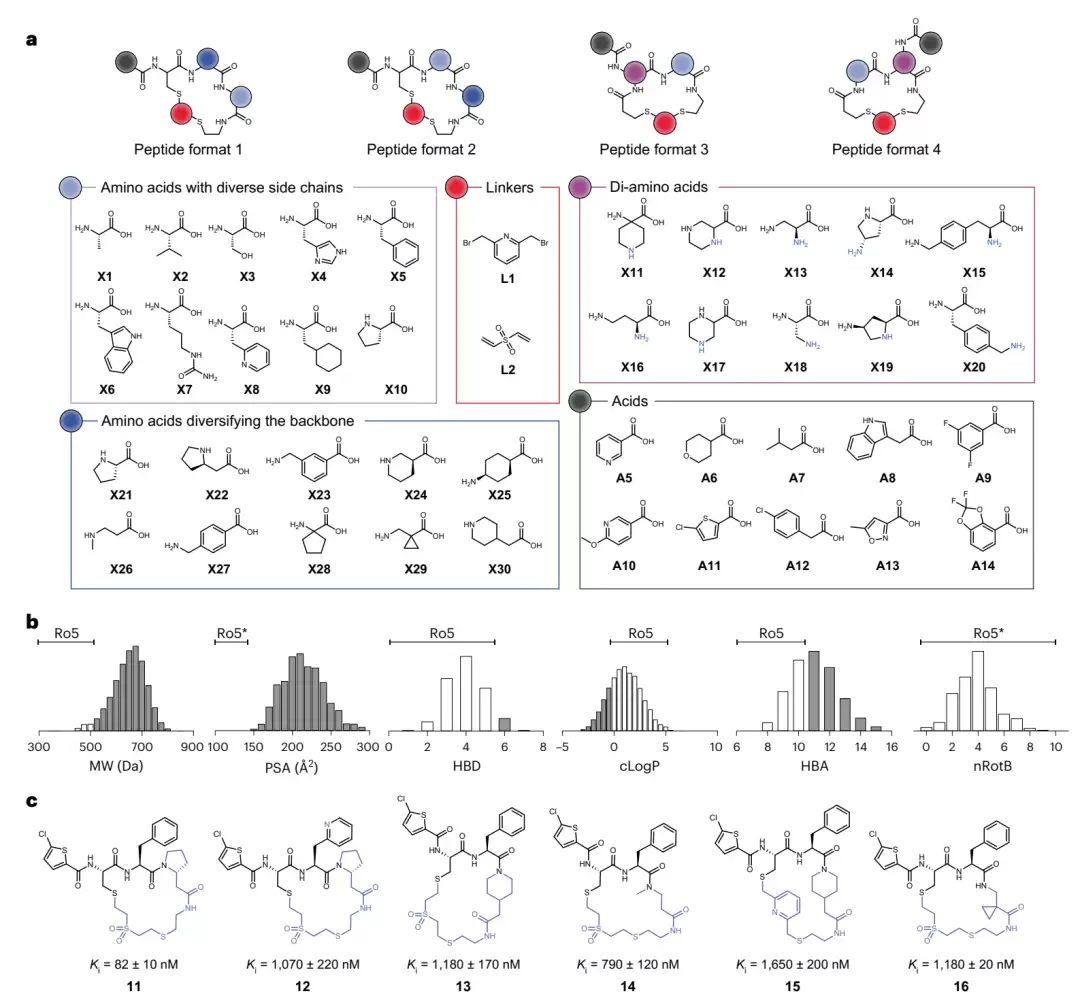 8,448 个环肽库的构成与凝血酶筛选结果。图 a 列出了建库所用的全部积木:十个侧链各异的氨基酸、十个改变骨架的氨基酸、十个二氨基酸、两个连接子、十个羧酸,以及四种肽的格式。图 b 以直方图展示整个文库的物化性质(分子量、极性表面积、氢键供体等),灰色部分表示超出“五规则”的肽,可见绝大多数都越界。图 c 展示了第一组六个最强命中肽(11–16)的结构及其各自的 Ki 值,共同骨架以黑色标出。
8,448 个环肽库的构成与凝血酶筛选结果。图 a 列出了建库所用的全部积木:十个侧链各异的氨基酸、十个改变骨架的氨基酸、十个二氨基酸、两个连接子、十个羧酸,以及四种肽的格式。图 b 以直方图展示整个文库的物化性质(分子量、极性表面积、氢键供体等),灰色部分表示超出“五规则”的肽,可见绝大多数都越界。图 c 展示了第一组六个最强命中肽(11–16)的结构及其各自的 Ki 值,共同骨架以黑色标出。
8,448 个环肽库的构成与凝血酶筛选结果。图 a 列出了建库所用的全部积木:十个侧链各异的氨基酸、十个改变骨架的氨基酸、十个二氨基酸、两个连接子、十个羧酸,以及四种肽的格式。图 b 以直方图展示整个文库的物化性质(分子量、极性表面积、氢键供体等),灰色部分表示超出“五规则”的肽,可见绝大多数都越界。图 c 展示了第一组六个最强命中肽(11–16)的结构及其各自的 Ki 值,共同骨架以黑色标出。
没有一个肽能三项全优
找到高亲和力的结合肽只是第一步。能否口服,还需考察另外三项体外指标:抗蛋白酶能力、细胞膜穿透性以及在肝脏中的清除速率。团队将 20 个肽(11–30)逐一进行了检测。
抗蛋白酶实验:将肽分别浸泡在模拟胃液(pH 1.2 的胃蛋白酶)和模拟肠液(pH 6.8 的胰酶)中,37°C 孵育 8 小时。大多数肽保持稳定——团队将此归因于它们主要由非天然氨基酸组成、分子量小、构象不易松动。
膜穿透性实验:采用 PAMPA 人工膜系统测定被动扩散能力。20 个肽的穿透性差异极大:有的能快速扩散,有的几乎纹丝不动。换算为表观渗透系数(logPapp),范围从几乎不穿透的约 −8 到完全穿透的约 −5,表现最好的六个肽在 −6.0 到 −5.2 之间,已接近参照药物华法林(−5.3)。
代谢稳定性实验:仍使用大鼠肝微粒体。结果显示,只有五个肽的清除速率慢于维拉帕米——团队将维拉帕米(一个清除偏快的口服药)定为可接受的稳定性下限。
将三项结果放在一起审视,结论非常清晰:每一项中都有几个肽表现不错,但没有任何一个肽能在三项指标上全部达标。
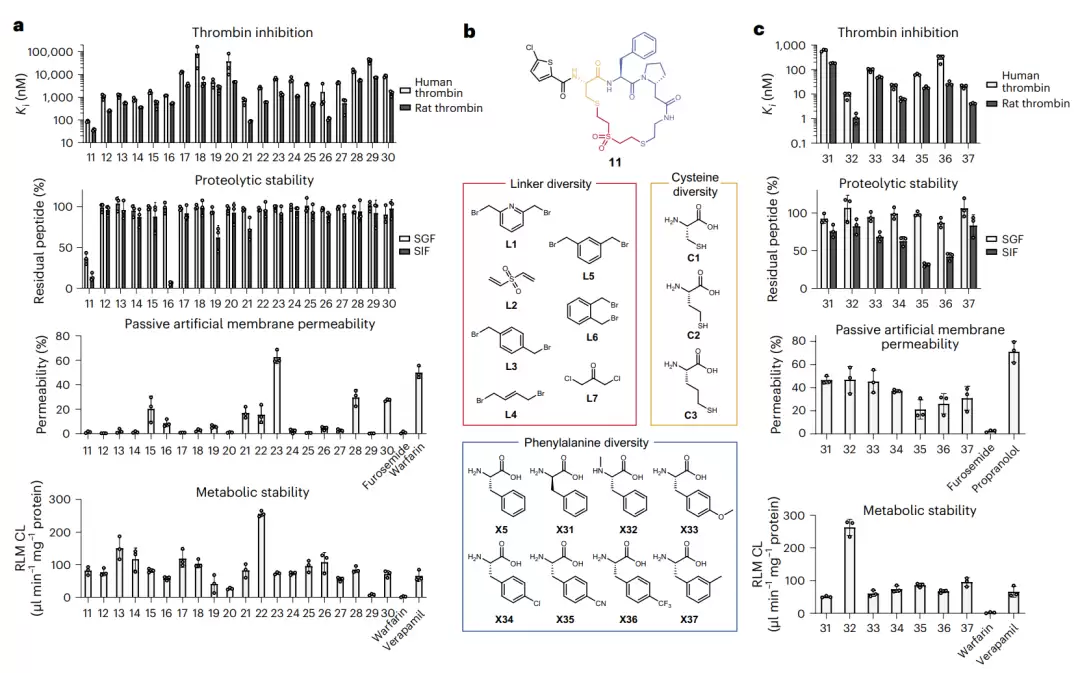 凝血酶抑制肽的体外表征与子库筛选。图 a 将肽 11–30 的四项指标(对人/大鼠凝血酶的抑制、抗胃肠液降解、人工膜渗透、肝微粒体代谢稳定)逐一列出,可直观看出没有哪个肽四项皆优。图 b 展示了基于环肽 11 构建子库时,在苯丙氨酸、连接子、半胱氨酸三个位点上用于替换的积木。图 c 展示了优化后一批肽(31–37)的同样四项表征数据。
凝血酶抑制肽的体外表征与子库筛选。图 a 将肽 11–30 的四项指标(对人/大鼠凝血酶的抑制、抗胃肠液降解、人工膜渗透、肝微粒体代谢稳定)逐一列出,可直观看出没有哪个肽四项皆优。图 b 展示了基于环肽 11 构建子库时,在苯丙氨酸、连接子、半胱氨酸三个位点上用于替换的积木。图 c 展示了优化后一批肽(31–37)的同样四项表征数据。
凝血酶抑制肽的体外表征与子库筛选。图 a 将肽 11–30 的四项指标(对人/大鼠凝血酶的抑制、抗胃肠液降解、人工膜渗透、肝微粒体代谢稳定)逐一列出,可直观看出没有哪个肽四项皆优。图 b 展示了基于环肽 11 构建子库时,在苯丙氨酸、连接子、半胱氨酸三个位点上用于替换的积木。图 c 展示了优化后一批肽(31–37)的同样四项表征数据。
一轮一轮,补上短板
既然没有现成的完美分子,团队便从最强的肽 11 出发,进行多轮迭代优化。高效的合成方法使得这种小规模快速测试几乎零成本。
第一轮,团队保留肽 11 中的 β-高脯氨酸和氯噻吩不变,仅在另外三个位置(半胱氨酸、苯丙氨酸、连接子)上更换不同的积木,构建了一个包含 168 个成员的子库(8 种苯丙氨酸 × 7 种连接子 × 3 种半胱氨酸)。这一轮还采用了一个巧妙的设计:将粗反应混合物直接加入 PAMPA 板的供体孔中,然后同时测定供体孔和受体孔中的凝血酶抑制活性。
读数表明:更换苯丙氨酸会大幅削弱活性,但替换半胱氨酸侧链和连接子仍可接受;受体孔数据显示,使用 L1 和 L4 连接子环化的肽,其膜穿透性明显优于原先使用砜基连接子 L2 的肽,其中 L4 在活性和穿透性方面均表现最佳。从挑出纯化的肽 31–37 中,最强的是肽 32:对人及大鼠凝血酶的 Ki 分别为 8.9 和 1.10 纳摩尔,比母体肽 11 强十倍。该肽抗蛋白酶、能穿透细胞膜,几乎全部达标,仅有一项例外:肽 32 在肝微粒体中清除速度异常快(260 微升/分钟/毫克),代谢稳定性不足,无法进入体内实验。
问题再次回到硫醚键。对肽 32 进行质谱分析,发现随着孵育时间增加,出现 +16 和 +32 的质量峰,指向硫醚键被氧化,很可能生成了亚砜。既然硫原子是软肋,干脆将其全部去除。第二轮,团队采用全碳氢连接子重新环化肽 32,依靠固相上的关环复分解(RCM)完成。
在七个通过 RCM 环化的肽 38–44 中,表现最佳的是肽 43:对人及大鼠凝血酶的 Ki 分别为 50 和 11.1 纳摩尔,代谢稳定性较肽 32 提高了三倍(清除率 85),且仍具备膜穿透能力。至此,肽 43 终于达到了进入体内实验的条件。
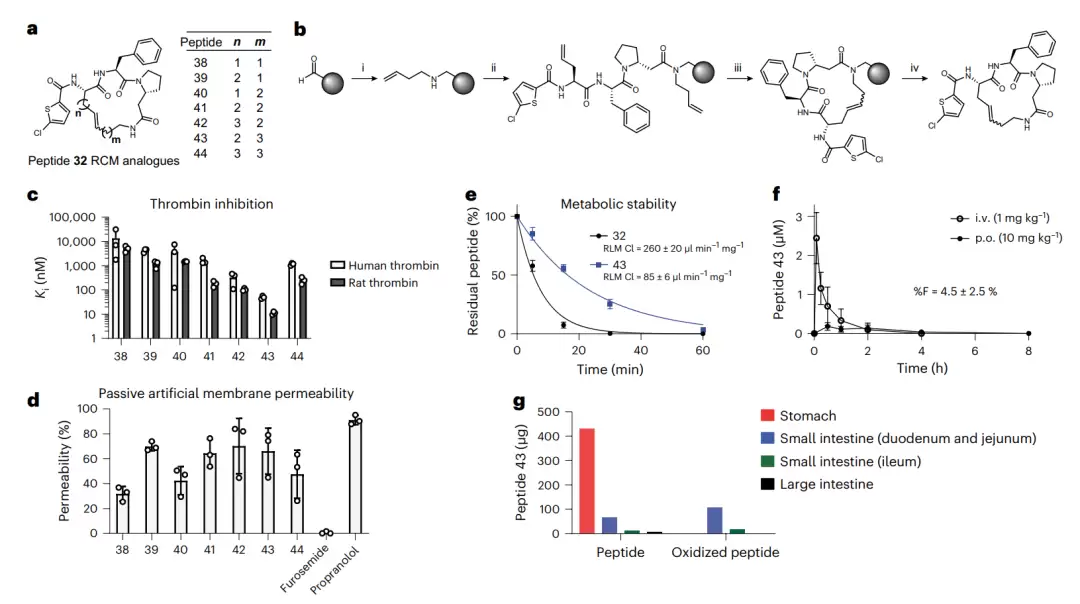 采用碳氢连接子环化的肽(基于肽 32)。图 a 展示了七个通过 RCM 环化的肽 38–44 的结构,连接子长度各不相同。图 b 描绘了 RCM 在固相上的合成步骤。图 e 将肽 32 与其 RCM 类似物 43 在肝微粒体中的稳定性进行对比,43 明显更稳定。图 f 展示了肽 43 在大鼠体内的口服与静脉血药曲线,图 g 显示口服后肽 43 在胃肠道各段大部分保持完整,但已能检测到少量被氧化的产物。
采用碳氢连接子环化的肽(基于肽 32)。图 a 展示了七个通过 RCM 环化的肽 38–44 的结构,连接子长度各不相同。图 b 描绘了 RCM 在固相上的合成步骤。图 e 将肽 32 与其 RCM 类似物 43 在肝微粒体中的稳定性进行对比,43 明显更稳定。图 f 展示了肽 43 在大鼠体内的口服与静脉血药曲线,图 g 显示口服后肽 43 在胃肠道各段大部分保持完整,但已能检测到少量被氧化的产物。
采用碳氢连接子环化的肽(基于肽 32)。图 a 展示了七个通过 RCM 环化的肽 38–44 的结构,连接子长度各不相同。图 b 描绘了 RCM 在固相上的合成步骤。图 e 将肽 32 与其 RCM 类似物 43 在肝微粒体中的稳定性进行对比,43 明显更稳定。图 f 展示了肽 43 在大鼠体内的口服与静脉血药曲线,图 g 显示口服后肽 43 在胃肠道各段大部分保持完整,但已能检测到少量被氧化的产物。
灌进大鼠的胃里
将肽 43 灌入大鼠胃中(n=3),血样分析表明它确实进入了血液循环:30 分钟后血浆峰浓度为 183 纳摩尔,口服生物利用度为 4.5%。作为阳性对照,他们使用了 Lokey 实验室设计、专门具备高膜穿透性的环六肽 1NMe3,该肽在相同条件下的口服生物利用度为 27%。
4.5% 仍不够理想。为了弄清肽 43 在体内的遭遇,团队再次给大鼠灌药,30 分钟后取出胃、小肠(十二指肠和空肠、回肠分开)及大肠分别分析。结果显示大部分肽保持完整,但在消化道中已能发现一些被氧化的肽(多出 16、32 的质量)。这提示只要进一步封堵残余的氧化位点,生物利用度还有提升空间。
第三轮的目标是消除肽 43 上最后一个氧化位点。质谱未能精确定位氧化发生的位置(找不到被氧化的碎片),但团队怀疑是氯噻吩基团。于是他们又进行了一轮快速替换:固定环状结构部分,仅在外围氨基上更换不同的羧酸。考虑到肽 43 很可能结合在凝血酶的 S1 口袋,他们挑选了 17 个同样能塞入 S1 口袋的羧酸进行测试,以氯噻吩作为阳性对照。
筛选出的肽 45 和 46 活性均与母体肽 43 相当。其中活性略强的肽 46,用 4-氯苯甲酸替换了氯噻吩,对人及大鼠凝血酶的 Ki 分别为 65 和 10.3 纳摩尔,抗蛋白酶和膜穿透性均良好,在肝微粒体中的稳定性又比肽 43 提升约一倍。专一性测试表明,肽 46 对凝血酶具有选择性抑制(对其他几种胰蛋白酶样蛋白酶基本无影响),且属于竞争性抑制剂。最后测试口服效果:肽 46 的血浆峰浓度达到 400 纳摩尔,是肽 43 的两倍;口服生物利用度升至 18.4%,已接近专为穿透设计造的参照肽 1NMe3 的 27%。
从肽 11 到肽 32(效力提高十倍),到肽 43(替换硫醚、可进入体内),再到肽 46(封堵最后氧化位点、口服生物利用度达 18%),这条优化路径上的每一步,都精准针对上一轮暴露出的短板。
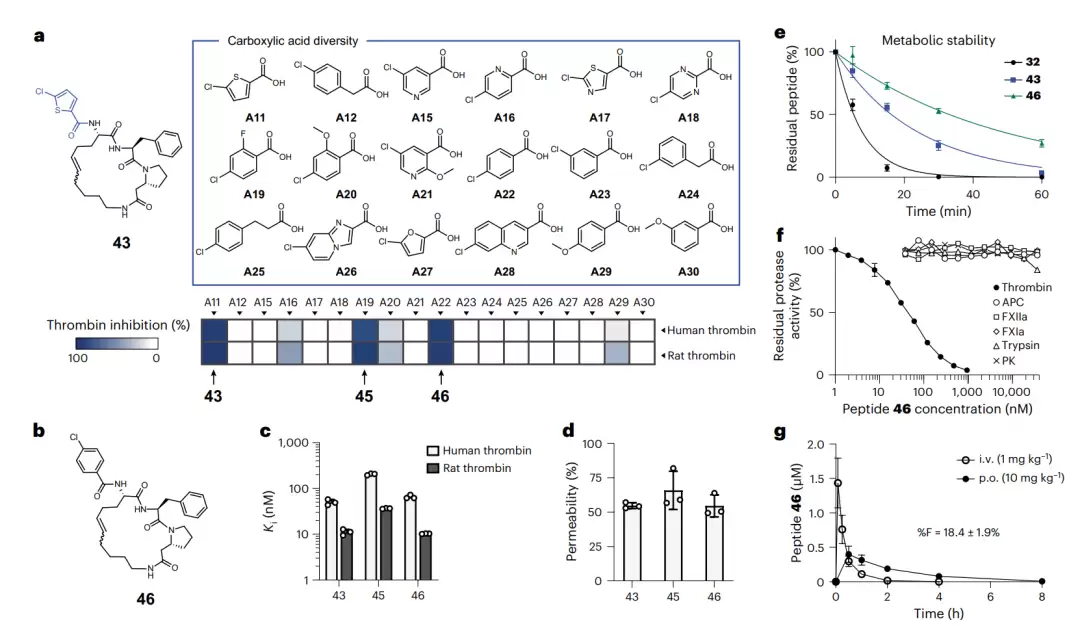 代谢更稳定、口服效果更佳的环肽 46。图 a 展示了基于肽 43、将外围氯噻吩替换为 17 种类似羧酸的子库,下方热图为这些肽对人及大鼠凝血酶的抑制强度。图 b 为肽 46 的化学结构。图 e 将肽 32、43、46 三者在肝微粒体中的稳定性进行对比,可看出逐代改善。图 f 为肽 46 的专一性谱,仅凝血酶被显著抑制。图 g 为肽 46 在大鼠体内的口服与静脉血药曲线,口服生物利用度为 18.4%。
代谢更稳定、口服效果更佳的环肽 46。图 a 展示了基于肽 43、将外围氯噻吩替换为 17 种类似羧酸的子库,下方热图为这些肽对人及大鼠凝血酶的抑制强度。图 b 为肽 46 的化学结构。图 e 将肽 32、43、46 三者在肝微粒体中的稳定性进行对比,可看出逐代改善。图 f 为肽 46 的专一性谱,仅凝血酶被显著抑制。图 g 为肽 46 在大鼠体内的口服与静脉血药曲线,口服生物利用度为 18.4%。
代谢更稳定、口服效果更佳的环肽 46。图 a 展示了基于肽 43、将外围氯噻吩替换为 17 种类似羧酸的子库,下方热图为这些肽对人及大鼠凝血酶的抑制强度。图 b 为肽 46 的化学结构。图 e 将肽 32、43、46 三者在肝微粒体中的稳定性进行对比,可看出逐代改善。图 f 为肽 46 的专一性谱,仅凝血酶被显著抑制。图 g 为肽 46 在大鼠体内的口服与静脉血药曲线,口服生物利用度为 18.4%。
这套方法的能效与局限
这项工作的真正成果,不仅是获得了一个口服生物利用度达 18% 的凝血酶抑制肽,更是一套可反复使用的肽类药物开发方法:利用硫醚环化构建可直接筛选的大规模文库,借助声波分液技术合成成千上万个环肽,再通过能同时评估结合与膜穿透能力的子库筛选,逐步将候选肽推向口服化。这套方法适用于任何具有功能读数的靶点。
但作者也明确指出了其局限性。
〔边界〕第一,硫醚键是否适合直接成药,作者坦言目前尚不确定。一方面,几种已上市的口服药本身含有硫醚键,他们的一些测试肽稳定性甚至超过了这些药物;另一方面,他们最强的几个肽恰好卡在硫醚氧化问题上,不得不替换。结论是稳定性很可能取决于具体序列,需要逐例分析。一个更彻底的策略是从一开始就采用 RCM 的全碳环,省去后续更换连接子的麻烦——但要使 RCM 能够高效环化大量不同肽段,仍需要大量技术开发。
第二,该肽目前还不能作为药物使用。要达到药理活性所需的亚纳摩尔级亲和力还有差距;作者所用均为市售构件,认为通过定制构件有望进一步提升。此外,RCM 环上的碳碳双键几何构型尚未确定,肽 46 本身即为 E/Z 异构体的混合物。
第三,目前所有的体内证据均来自大鼠,且靶点仅涉及凝血酶。方法的普适性仍停留在理论论证与单个成功案例层面,尚未在更多靶点——尤其是更具挑战性的靶点(如蛋白-蛋白相互作用)上得到验证。
团队特别强调了不带电的设计理念。大多数凝血酶抑制剂需要依靠带电基团结合 S1 口袋,而带电几乎注定难以口服;这批肽则完全不带电,从源头上绕开了这一难题——这也是它们能比达比加群酯实现更好口服效果的根本原因。
将注射剂变为药片
很长一段时间里,肽类药物被困在尴尬的境地:它们结合靶点的能力一流,却几乎必须通过注射给药。这项研究并未声称解决了所有问题——所制备的肽距离真正的药物尚有距离,证据也仅限于大鼠和单一靶点。但它成功将一件原本依赖运气和大量手工劳动才能偶然达成的事情,转化为一个可逐步逼近目标的过程:每暴露一个短板,就有方法去修补;每修补一次,就离口服更近一步。
如果这套方法的简便与稳健性能如作者期望的那样被广泛应用,那么那些至今只能靠注射、甚至无药可用的棘手靶点,或许终有一天会等来一颗可直接吞服的小环肽。将注射剂变为药片,其意义远不止于便利——它决定了一种药物能否真正走进那些原本无法触及它的人们的生活。
参考文献
原文 De novo development of small cyclic peptides that are orally bioa vailable(Merz、Habeshian、Li 等,通讯作者 Christian Heinis,EPFL),发表于《Nature Chemical Biology》2024 年第 20 卷 624–633 页,DOI 10.1038/s41589-023-01496-y。
本文为开放获取(CC BY 4.0),原始数据随论文一并提供;利益声明中,通讯作者等为衍生公司 Orbis Medicines 的创始人。


游乐网为非赢利性网站,所展示的游戏/软件/文章内容均来自于互联网或第三方用户上传分享,版权归原作者所有,本站不承担相应法律责任。如您发现有涉嫌抄袭侵权的内容,请联系youleyoucom@outlook.com。
 同类文章
同类文章

Windows Docker Desktop RabbitMQ生产级部署完整指南
前言 在 Windows 本地开发环境中,直接安装 RabbitMQ 确实颇为周折:需要单独配置 Erlang 运行环境、手动管理环境变量、服务启停全凭手工操作。更令人困扰的是,版本兼容冲突、端口占用、环境不一致等问题层出不穷。笔者见过不少开发者为搭建环境就得耗费整整半天时间。 相比之下,借助 Do

AI搜索重构制造业采购逻辑的阿里云企业级GEOCMS优化实践
先分享一个切实感受。过去两年,我们与福建制造企业合作较为频繁,发现一个非常突出的现象:超过80%的企业官网,产品参数仍然存放在PDF或图片中。AI爬虫?根本无法抓取。这些企业技术实力不弱、资质证照齐全、应用案例也丰富,但在AI搜索这一全新战场上,它们几乎处于隐身状态。 一、一个正在发生的行业变化 A

阿里云Token Plan团队版功能价格与省钱购买指南
阿里云百炼近期推出了名为“Token Plan 团队版”的全新服务,这一服务专为企业与开发者量身打造,定位为AI大模型订阅平台。通过引入Credits作为统一计量单位,将文本生成、图像生成等多模态AI能力纳入单一计费体系,同时无缝兼容主流AI编程工具及智能体(Agent)生态系统。其核心亮点包括:全

阿里云物联网.NET Core客户端位置信息上报
阿里云物联网平台的位置服务并非一个完全独立的功能模块。位置信息可包含二维坐标与三维坐标,而位置数据的来源本质上是借助设备属性进行上传。换言之,若要让设备上报位置,您需先将其视为一个普通属性进行处理。 1)添加二维位置数据 操作过程十分简洁。进入数据分析 → 空间数据可视化 → 二维数据,点击添加,将

年阿里云服务器选型配置与网站部署全攻略
2026年,阿里云服务器生态已高度成熟,形成了清晰的轻量应用服务器与ECS云服务器两大产品阵营。无论你是计划搭建个人博客、企业官网,还是运营电商平台、进行应用开发,基本都能找到理想的解决方案。本指南将从服务器选型、配置选择、部署流程到安全运维,系统梳理2026年最实用的操作要点,帮助你少走弯路,让网








- 日榜
- 周榜
- 月榜



 相关攻略
相关攻略
 2026-06-29 17:42
2026-06-29 17:42
2026-06-29 17:42
2026-06-29 17:42
2026-06-29 17:41
2026-06-29 17:41
2026-06-29 17:41
2026-06-29 17:41
热门教程
2026-06-29 17:42
2026-06-29 17:42
2026-06-29 17:42
2026-06-29 17:42
2026-06-29 17:41
2026-06-29 17:41
2026-06-29 17:41
2026-06-29 17:41
热门教程
- 游戏攻略
- 安卓教程
- 苹果教程
- 电脑教程















 热门话题
热门话题
