




LiteFold发布TB级蛋白数据,助力蛋白折叠研究


近年来,蛋白质人工智能的演进可以用一句话概括:模型性能不断增强。
AlphaFold 将蛋白质结构预测从少数结构生物学家的专属技能,转变为许多实验室都能轻松使用的工具。ESM 等蛋白质语言模型将序列、进化与结构表征的能力推向了更大规模。在蛋白质设计、突变效应预测、稳定性建模、抗体与多肽工程等领域,深度学习模型已全面融入工作流程。
然而,当研究者真正动手训练模型、复现实验或搭建基准测试时,深入探索后会发现,问题往往并非出在模型本身,而是数据层面先卡住了。
蛋白质数据从来不是一份干净规整的表格。它们可能隐匿于论文的补充材料中,存放在某个 FTP 目录下,或者分散在 UniProt、PDB、AlphaFold DB、ESM Atlas、ProteinGym、MegaScale、Pfam、InterPro、GOA 等多个来源之间。一个数据源按序列组织,另一个按结构链组织;一个分数越大越好,另一个越小越稳定;一个记录的是突变效应,另一个只存储结构坐标;一个有许可说明,另一个还需要追溯上游版本。
因此,当 LiteFold 发布 AminoWeb 时,它真正切入的并非又一个新模型,而是一个更基础、也更棘手的难题:既然蛋白质 AI 已经迈入大模型时代,那么支撑它的数据层,是否也应该像机器学习基础设施那样被认真搭建起来?

它的核心功能是什么?
AminoWeb 是 LiteFold 在 Hugging Face 上发布的一套蛋白质机器学习数据集合。根据发布信息,它整合了 29 个经过清洗、专门面向机器学习整理的数据集,总规模约为 7.56 TB;Hugging Face collection 页面则将其概括为约 8 TB 的蛋白质数据集合。
覆盖范围并非单一任务,而是一组蛋白质机器学习中最常见的输入与信号:序列、结构、功能注释、多序列比对、突变效应、稳定性、结合与相互作用等。
换句话说,它不是来提出新的结构预测模型,也不是提供一套全新的药物设计流程。它更像是将蛋白质 AI 中那些分散的数据源,重新整理成模型工程师更容易读取、检索、训练和复现实验的形式。
这项工作看似没有发布新模型那般耀眼,但它解决的是一个更日常、也更磨人的问题:数据能否稳定地读入,能否知道每一行的来源,能否避免训练与测试之间因同源蛋白泄漏而虚高,能否让不同任务之间重复利用。
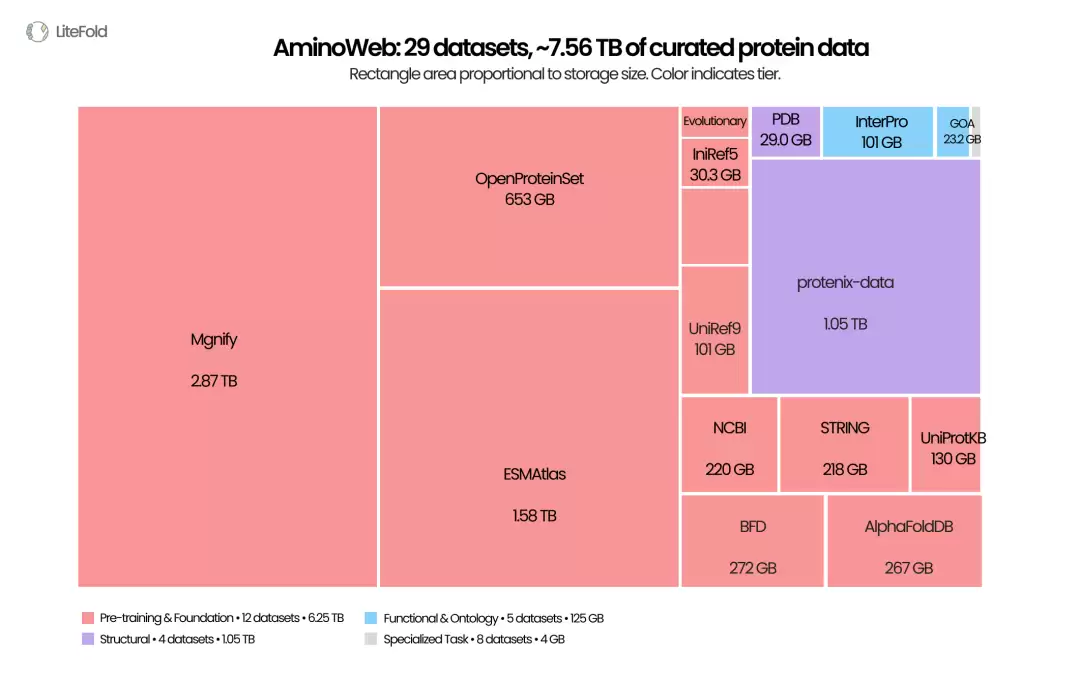 AminoWeb 将蛋白质的序列、结构、功能、突变效应、稳定性与相互作用等多维数据整合为可便捷访问的数据集合。图中面积代表数据体量,不直接反映数据质量或任务权重。
AminoWeb 将蛋白质的序列、结构、功能、突变效应、稳定性与相互作用等多维数据整合为可便捷访问的数据集合。图中面积代表数据体量,不直接反映数据质量或任务权重。
数据层为何成为瓶颈?
大语言模型领域早已习惯将数据集视为基础设施进行讨论。FineWeb、The Pile、RedPajama、Dolma 这些名字,本质上不只是数据本身,它们代表了语料收集、清洗、去重、记录来源与服务模型训练的整体工程实践。
蛋白质机器学习的问题则复杂得多。
自然语言中,两段网页文本之间固然可能存在重复与污染,但蛋白质数据中存在一种更隐蔽的重复:同源性。两个蛋白序列可能不完全相同,但它们可能来自共同祖先,结构与功能高度相关。如果训练集和测试集中混入了高度相似的同源蛋白,模型表面看起来泛化良好,实际可能只是学会了相近家族的“套路”,并未真正掌握举一反三的能力。
这也是蛋白质基准测试经常令人头疼的原因。随机划分虽然便捷,却未必能模拟真实的发现任务。真正有挑战的场景,往往是让模型面对远离训练分布的新家族、新结构域、新突变组合,甚至是一些实验注释稀少的序列空间。
另一个难点在于分数的“口径”问题。
突变效应数据、稳定性数据、结合数据、功能注释数据,看起来都能转化为监督学习任务,但它们的分数含义各不相同。有的分数表示活性升高,有的表示适应性下降,有的代表 ΔΔG,有的来自高通量实验,有的源自精心整理的数据库。如果为了统一格式而粗暴地将它们全部转为同一方向,反而可能洗掉原始的生物学含义。
因此,整理蛋白质数据绝不只是简单地将 CSV 转为 Parquet。更关键的是,保留原始信息的同时,确保模型工程能够稳定地使用这些数据。
AminoWeb 整合了哪些内容?
从 collection 页面来看,AminoWeb 覆盖了多类常用的蛋白质数据源,大致可分为以下几组。
数据类型 | 代表数据源 | 对模型训练的价值 | 使用注意事项 |
|---|---|---|---|
序列与同源信息 | UniProtKB、UniRef50、UniRef90、BFD、MGnify、NCBI | 训练蛋白语言模型,构建多序列比对,执行序列检索与家族分析 | 序列冗余度高,划分时需控制同源泄漏 |
结构数据 | PDB、AlphaFoldDB、ESMAtlas、CATH | 结构预测、结构表征学习、结构检索、结构条件设计 | PDB 为实验结构,AlphaFoldDB 和 ESMAtlas 为预测结构,不可混为一谈 |
功能与注释 | GO、GOA、InterPro、Pfam、Human Protein Atlas | 功能预测、结构域识别、蛋白家族建模、多任务学习 | 注释密度分布不均,热门蛋白与模式物种可能过度代表 |
突变效应与稳定性 | ProteinGym、MegaScale、FireProtDB、FLIP2 | 突变效应预测、蛋白稳定性建模、序列优化 | 不同实验体系的分数方向与噪声水平各异 |
结合与相互作用 | SKEMPI2、IEDB、STRING | 蛋白互作、抗原表位识别、结合变化预测 | 数据来源混合,需区分实验置信度与计算预测 |
化学与特殊残基 | PDB-CCD、SwissSidechain、CycPepMPDB | 处理配体、非天然氨基酸、环肽等非标准对象 | 对药物发现很重要,但标准蛋白模型未必天然支持 |
(为方便阅读,将 collection 中可见的数据源按常见蛋白质机器学习任务重新归类。)
这张表基本勾勒出 AminoWeb 的定位。
它尝试将蛋白质 AI 的几个关键数据面纳入同一入口:序列空间、结构空间、功能注释、实验测量、相互作用以及非标准化学组件。
对模型开发者而言,这意味着一个实际的好处:无需每次从不同数据库重新编写下载、解析和字段映射脚本。至少在起步阶段,可以用统一的数据入口快速搭建任务。
传统方案为何不够?
过去做蛋白质机器学习,常见方式是每篇论文各自准备一份数据。
在研究规模较小时,这并非问题。研究者可以下载论文的补充表格,编写脚本清理突变格式,再按论文定义的划分方式拆分训练集和测试集。但一旦模型与任务规模扩大,问题便暴露无遗。
第一,清洗过程难以复现。
处理同一数据源时,是否去掉缺失值、是否合并重复突变、是否保留低置信度结构、是否统一 isoform、是否筛掉过长序列……这些决策都会影响结果。论文通常不会将每一步工程细节写得足够详尽。
第二,跨任务复用困难。
一个模型可能希望同时使用序列、结构、功能、突变效应和稳定性数据。每个数据源都有自己的主键与字段习惯:UniProt accession、PDB chain、AlphaFold ID、MGnify ID、突变 notation、结构域坐标……若缺乏统一映射,后续建模将消耗大量“脏活累活”。
第三,评测易受数据划分方式影响。
蛋白质任务尤其担心训练集与测试集之间存在高度相似的同源蛋白。一个模型在随机划分上表现良好,并不代表它真正掌握了远距离泛化能力。很多时候,基准测试的可信度取决于数据划分的质量,而不仅仅是模型效果。
这也是 AminoWeb 特意强调 homology-aware splits 的原因——它指向的不只是格式问题,而是评测可信度的根本问题。
新设计解决了哪些问题?
AminoWeb 的核心价值可概括为四个层面。
第一层:格式。
数据被整理成更适合机器学习读取的形式,例如 Parquet、JSONL、WebDataset 等。Parquet 对大表、列式读取和类型约束更友好;JSONL 便于保留原始行信息;WebDataset 则更适合承载大规模结构文件或分片数据。
这一步听起来偏工程化,但它直接影响使用门槛。对于 TB 级别的数据,能否 stream、能否只读部分字段、能否按 shard 下载,差别可谓天差地别。
第二层:来源追踪。
在多个 dataset card 中,可以看到类似 dataset_id、source_file、row_index、raw upstream row 的设计。它们的意义在于:模型读到一行数据时,理论上可以追溯它来自哪个上游文件、哪一行、原始字段是什么。
这对科学数据尤为重要。因为蛋白质数据库并非静态,版本会更新,字段会变化,注释会修订。若无 provenance,模型结果一旦出现问题,很难追查到源头。
第三层:分数口径。
AminoWeb 发布信息中特别提到 preserved score conventions,即保留原始分数约定。这个决定很关键。
许多人在整理数据集时倾向于将所有分数转为统一方向(比如“越大越好”)。但在蛋白质实验数据中,这样做可能抹掉上游实验语境。更稳妥的做法是保留原始分数,同时在 dataset card 或字段说明中告知用户如何解释。
第四层:评测划分。
蛋白质任务的划分不能只看样本随机性,还要考虑序列、结构与家族关系。AminoWeb 将 homology-aware splits 作为发布亮点之一,说明它已意识到蛋白质 ML 的核心风险之一:同源泄漏。
当然,也需要理性看待:并非所有子数据集都天然具备同一种划分方式。具体到某个任务,仍需查看对应的 dataset card,确认划分规则、字段含义与适用边界。
结果到底说明了什么?
从本次发布可以看出几个信号。
AlphaFoldDB 在 AminoWeb 中被整理为 prediction index,数据页显示约 2.47 亿行,并划分为 train 与 test。需要注意,它更像是一个预测结构索引与元数据表,而非简单等同于完整的结构坐标仓库。
ESMAtlas 数据页显示为大规模 WebDataset 镜像,文件体量约 1.74 TB,并保留了结构文件、metadata、confidence scores 等信息。它还明确提醒:ESMAtlas 结构为计算预测,并非实验结构,下游任务需借助 pLDDT、pTM 等置信度指标进行筛选。
ProteinGym 数据页显示约 293 万行,面向蛋白适应度与突变效应预测,是评估蛋白语言模型、逆折叠模型和监督预测模型的重要基准。
MegaScale-Tsuboyama2023 数据页显示约 29.9 万行整理记录,对应大规模蛋白折叠稳定性实验数据。它的价值在于将结构之外的热力学稳定性信号也带入了模型训练与评估。
Pfam 数据页显示约 1.28 亿行,主要承载蛋白家族与结构域区域注释。对于功能预测、结构域识别和蛋白家族学习等任务,这类注释数据比单纯的序列更接近可解释的生物学标签。
这些数字不应被简单理解为“越大越好”。更准确的理解是:AminoWeb 将不同粒度的蛋白质数据,放到了一个相对统一的机器学习入口下,让研究者能更快地构造跨数据源的任务。
但它并非万能。
预测结构终究是预测结构。低置信度区域、无序区、复合物界面、构象变化与活性位点几何,都需要谨慎对待。功能注释仍存在偏倚。突变效应数据依然受实验体系影响。稳定性数据与结合数据也不能直接互相替代。
数据被整理好,并不代表生物学问题就自动解决了。
对蛋白质 AI 而言,它的意义何在?
AminoWeb 的意义不在于让某个模型立刻变强,而在于将蛋白质 AI 的一个长期瓶颈显性化了:模型规模扩张以后,数据层也必须进入工程化阶段。
这将影响几类工作。
对基础模型训练来说,统一的数据入口可以降低预训练、微调和多任务学习的工程成本。序列、结构、MSA、功能注释、突变效应和稳定性数据若能更容易组合,模型就更容易从单一任务走向多模态蛋白质表征。
对基准测试而言,来源追踪与划分规则将变得更加重要。未来评价蛋白质模型,不能只看指标,还要看测试集与训练集之间的同源关系、数据版本、标签来源与任务定义。
对药物发现与蛋白工程来说,AminoWeb 这类数据集合可能让更多团队快速搭建原型。例如先用 ProteinGym 做突变效应评估,再结合结构数据与稳定性数据筛选候选,最后将相互作用或表位数据接入后续分析。它不会替代实验,但能让计算筛选的流程更可复现。
对开源生态来说,这也可能推动蛋白质 AI 从“论文数据”走向“数据资产”。一个数据集不再只是一个下载链接,而是要有字段、版本、许可、来源、拆分、使用示例与限制说明。
这件事听起来或许不如发布一个高调模型那般令人兴奋,但它可能更接近这个领域真正需要的底层基础设施。
参考文献
[1]. LiteFold. AminoWeb Collection. https://huggingface.co/collections/LiteFold/aminoweb
[2]. LiteFold

游乐网为非赢利性网站,所展示的游戏/软件/文章内容均来自于互联网或第三方用户上传分享,版权归原作者所有,本站不承担相应法律责任。如您发现有涉嫌抄袭侵权的内容,请联系youleyoucom@outlook.com。
 同类文章
同类文章

如何高效将多个表格数据合并汇总到一张表中
高效合并多个表格:Excel数据汇总的四大实用技巧 在日常办公中,我们常常需要面对这样的任务:几个分散在不同工作表中的表格,各自记录了不同的数据维度,但最终需要将它们整合到一起进行分析或制作报告。听起来有些繁琐,但Excel其实已经内置了多种成熟的解决方案。接下来,我将逐一拆解这四种方法,帮你找到最

免费AI智能写作生成器让你轻松解决创作难题
内容创作的模式,正被AI技术悄然重塑。如今,只需输入几个核心关键词,一篇结构完整的文章就能自动产出——这对内容创作者、市场营销人员和自媒体人而言,无疑是一大福音。写作过程往往耗时费力,而免费AI智能写作生成器的出现,恰好精准解决了这一难题。借助这些工具,创作者能够在更短的时间内产出更多优质内容,从而

A表格中快速提取B表格数据的高效方法
如何快速从A表格中提取B表格的数据提升工作效率的有效策略 在职场日常工作中,数据处理与分析几乎是每位从业者都要面对的必修课。尤其是当你需要从一个数据源(我们称之为A表格)中提取特定信息,用以填充另一个目标表格(即B表格)时,选对方法往往能让效率相差数倍。以下介绍的几种方法均经过实际工作验证,你可以根

如何高效选取Excel表格部分数据提升工作效率技巧
Excel表格部分数据选取全攻略:筛选、条件格式、公式与透视表 日常处理数据时,最令人困扰的往往不是数据本身,而是如何从一张包含成百上千行的大表中,迅速定位并提取出所需部分。无论是筛选特定记录,还是获取关键信息,若方法不当,仅靠鼠标拖拽就可能耗费大量时间。以下介绍的几种Excel数据选取技巧,均经过

中秋节放假通知模板 正式温暖关怀版
适合需求: 2023年中秋节放假通知模板 尊敬的公司全体员工: 中秋佳节即将来临,首先感谢大家近期在工作中的辛勤付出与坚守。每年中秋月圆之际,大家都期盼与家人团聚,共享温馨时刻。公司也衷心希望每一位员工都能在这个充满温情的传统节日里,与亲人欢聚一堂,共度美好时光。 依据公司统一安排,现将2023年中








- 日榜
- 周榜
- 月榜
 1
1
 2
2
 3
3
 4
5
6
7
8
9
10
相关攻略
4
5
6
7
8
9
10
相关攻略
 2015-03-10 11:25
2015-03-10 11:05
2021-08-04 13:30
2015-03-10 11:22
2015-03-10 12:39
2022-05-16 18:57
2025-05-23 13:43
2025-05-23 14:01
热门教程
2015-03-10 11:25
2015-03-10 11:05
2021-08-04 13:30
2015-03-10 11:22
2015-03-10 12:39
2022-05-16 18:57
2025-05-23 13:43
2025-05-23 14:01
热门教程
- 游戏攻略
- 安卓教程
- 苹果教程
- 电脑教程































 热门话题
热门话题
