




RAPIDS实现单细胞数据分析速度提升的实用技巧

RAPIDS-singlecell通过GPU加速单细胞RNA测序分析,利用cunData数据结构与预处理、聚类等模块,实现10倍以上性能提升。在90000和500000细胞数据集上,完整分析耗时从CPU的18分钟和41分钟分别缩短至51秒和110秒,显著加快研究迭代。
单细胞测序技术已成为生物医学研究领域最前沿的工具之一。它能够在单细胞层面揭示转录组与表观基因组的动态变化,为科学家带来全新的研究视角。然而,随着实验规模不断扩大,涉及超过百万个细胞的实验已逐渐成为常态,传统的基于 CPU 的分析流程在性能上面临巨大挑战。本教程将系统介绍如何利用 RAPIDS-singlecell 库,为单细胞 RNA 测序(scRNA-seq)分析引入 GPU 加速,实现 10 到 20 倍甚至更高的性能提升,从而显著加快研究迭代速度,让洞察更及时、分析更高效。
1. 为什么需要 GPU 加速?
单细胞分析流程高度依赖迭代计算,快速算法的引入能极大缩短分析周期。Scanpy 作为 scverse 生态系统中广泛使用的单细胞分析套件,其算法大多运行在 CPU 上,在处理大规模数据时速度明显下降。RAPIDS 和 CuPy 等 GPU 加速库的出现,为突破这一瓶颈提供了理想的解决方案。RAPIDS-singlecell 正是为此而生——它提供了与 Scanpy 几乎一一对应的 GPU 加速函数,无需深入学习 CUDA 编程,即可轻松享受 GPU 带来的极速运算体验。
2. RAPIDS-singlecell 生态系统概览
RAPIDS-singlecell 遵循与 scverse Python 库类似的可用性模型,采用 Python 编写,并将性能关键部分部署在 GPU 上。它主要由以下五类组件构成:
- cunData – GPU 端轻量化的 AnnData 对象
- 预处理函数 (cunData_funcs) – 加速过滤、标准化、PCA 等操作
- 工具 (scanpy_gpu) – 加速聚类、UMAP、t-SNE 等分析
- 解耦器 (decoupler_gpu) – 加速基因集与转录因子活性分析
- Squidpy 开发 – 加速空间分子数据分析
3. 核心数据结构:cunData
cunData 是 GPU 版的 AnnData 对象,它将计数矩阵 .X 存储为 CuPy 稀疏矩阵 并置于 GPU 上,从而大幅提升计算效率。同时,它还支持将多个版本的计数矩阵存储在 .layers 中(同样位于 GPU),有效减少数据迁移开销。
cunData 还包含细胞注释 (.obs)、基因注释 (.var)、非结构化注释 (.uns) 以及多维注释 (.obsm, .vrm)。切片操作与 AnnData 类似,但始终返回完整副本(而非视图)。
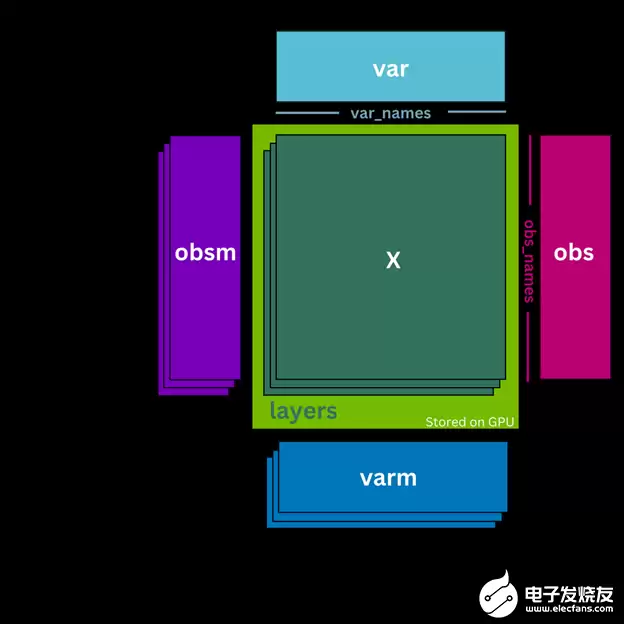 图 1. cunData 类结构示意图
图 1. cunData 类结构示意图
小提示: 建议在预处理阶段使用 cunData,分析完成后可通过 .to_AnnData() 转换回 AnnData 对象,以便利用 Scanpy 丰富的可视化功能。
代码示例:将 AnnData 转换为 cunData
import scanpy as sc
import rapids_singlecell as rsc
adata = sc.read("PATH TO DATASET")
cudata = rsc.cunnData.cunnData(adata=adata)
4. 预处理加速(cunData_funcs)
预处理函数存储在 cunData_funcs 模块中,为 Scanpy 的预处理功能提供 GPU 替代方案。所有操作直接在 cunData 对象上执行。
- 过滤细胞和基因:
filter_cells、filter_genes - 质量控制:
calculate_qc_metrics - 标准化:
normalize_total、log1p、normalize_pearson_residuals - 高变基因选择:
highly_variable_genes(支持 seurat、cellranger、seurat_v3、pearson_residuals、poisson_gene_selection 等多种方法) - 回归和缩放:
regress_out(使用 cuML 线性回归)、scale - 降维:
pca(支持 PCA、Truncated SVD、Incremental PCA,并可选指定 layer)
小提示: regress_out 支持多目标回归(cuML 22.12 引入),同时保持向后兼容。
代码示例:完整预处理流程
# Basic QC
rsc.pp.flag_gene_family(cudata, gene_family_name="MT", gene_family_prefix="mt-")
rsc.pp.calculate_qc_metrics(cudata, qc_vars=["MT"])
cudata = cudata[cudata.obs["n_genes_by_counts"] > 500]
cudata = cudata[cudata.obs["pct_counts_MT"] < 20]
rsc.pp.filter_genes(cudata, min_count=3)
# Log normalization and highly variable gene selection
cudata.layers["counts"] = cudata.X.copy()
rsc.pp.normalize_total(cudata, target_sum=1e4)
rsc.pp.log1p(cudata)
rsc.pp.highly_variable_genes(cudata, n_top_genes=5000, fla vor="seurat_v3", layer="counts")
cudata = cudata[:, cudata.var["highly_variable"] == True]
# Regression, scaling and PCA
rsc.pp.regress_out(cudata, keys=["total_counts", "pct_counts_MT"])
rsc.pp.scale(cudata, max_value=10)
rsc.pp.pca(cudata, n_comps=100)
sc.pl.pca_variance_ratio(cudata, log=True, n_pcs=100)
# Convert back to AnnData for downstream visualization
adata_preprocessed = cudata.to_AnnData()
5. 工具:scanpy_gpu
scanpy_gpu 模块提供在 AnnData 对象上运行的 GPU 加速函数,语法与 Scanpy 保持一致,元数据写入 .uns 属性。该模块扩展了 Scanpy 对 GPU 的支持,新增了以下功能:
- 聚类: Leiden 和 Louvain(基于 cuGraph)
- 可视化布局: Force Atlas 2
- 降维: PCA、diffusion maps(CuPy 实现)
- 密度估计: KDE(cuML)
- 批量校正: Harmony Integration(通过
harmony_gpu调用)
绘图可直接使用 Scanpy 的绘图函数,无缝集成 scverse 生态。
6. 解耦器(decoupler_gpu)
decoupler 工具提供统一的框架,用于分析生物活动(如基因集、转录因子活性)。decoupler_gpu 重新实现了加权和 (run_wsum) 和多元线性模型 (run_mlm),性能提升显著,例如 wsum 加速可达 37 倍。
7. Squidpy 开发
RAPIDS-singlecell 正在扩展对空间分子数据(Squidpy)的加速支持:
- 空间自相关: Morans I 和 Gearys C 加速 高达 100 倍
- 配体-受体交互分析 (ligrec): 加速 超过 10 倍
8. 基准测试:性能提升一览
以下三个表格展示了在不同硬件配置下,RAPIDS-singlecell 相较于传统 CPU 流程的加速效果。数据来源于 90000 细胞数据集和 500000 细胞数据集,分别在服务器节点(A100 80GB)和消费级桌面(RTX 3090)上进行测试。
表 1:90000 细胞(服务器节点:2×AMD Epyc Milan 7543 + 500GB RAM + A100 80GB)
| 作用 | CPU | GPU | 加速 |
| 整个笔记本(不包括 PR 功能) | 1106 秒(18.5 分钟) | 51 秒 | 21 倍 |
| 预处理 | 74 秒 | 8 秒 | 9 倍 |
| HVG (Seurat v3) | 27 秒 | 1.6 秒 | 16 倍 |
| Regress out | 35 秒 | 0.7 秒 | 50 倍 |
| scale | 3.2 秒 | 0.4 秒 | 8 倍 |
| PCA | 417 秒 | 18 秒 | 23 倍 |
| Neighbors | 22 秒 | 5.1 秒 | 4.3 倍 |
| UMAP | 36 秒 | 0.4 秒 | 90 倍 |
| TSNE | 133 秒 | 2.4 秒 | 55 倍 |
| Louvain | 17 秒 | 0.6 秒 | 28 倍 |
| Leiden | 14 秒 | 0.2 秒 | 70 倍 |
| 逻辑回归 | 58 秒 | 3.7 秒 | 15 倍 |
| 绘图(FA2) | 256 秒 | 0.3 秒 | 850 倍 |
| run_mlm (DoRothEA) | 83 秒 | 12 秒 | 7 倍 |
| Run_wsum (程序) | 970 秒(16 分钟) | 26 秒 | 37 倍 |
表 2:500000 细胞(服务器节点 A100 vs 消费级 RTX 3090)
| 作用 | CPU | GPU (A100) | GPU (3090) | 加速 |
| 整个笔记本 | 2460 秒(41 分钟) | 110 秒 | 290 秒 | 22 倍 |
| 预处理 | 305 秒 | 28 秒 | 169 秒 | 10 倍 |
| HVG (Seurat v3) | 48 秒 | 1.5 秒 | 13 秒 | 32 倍 |
| Regress out | 104 秒 | 5.1 秒 | 16 秒 | 20 倍 |
| scale | 8.4 秒 | 1.3 秒 | 5 秒 | 6.4 倍 |
| PCA | 86 秒 | 3.7 秒 | 35 秒 | 23 倍 |
| Neighbors | 74 秒 | 17.1 秒 | 18.3 秒 | 4.3 倍 |
| UMAP | 281 秒(4.6 分钟) | 6.7 秒 | 7.6 秒 | 60 倍 |
| TSNE | 786 秒(13 分钟) | 10 秒 | 12.9 秒 | 105 倍 |
| Louvain | 283 秒(4.5 分钟) | 4.5 秒 | 5.7 秒 | 62 倍 |
| Leiden | 282 秒(4.5 分钟) | 0.6 秒 | 0.9 秒 | 470 倍 |
| 逻辑回归 | 452 秒(7.5 分钟) | 33 秒 | 63 秒 | 13 倍 |
| 扩散贴图 | 30 秒 | 0.75 秒 | 1.3 秒 | 40 倍 |
| 重型车辆(公共车辆) | 104 秒 | 2.1 秒 | 15.6 秒 | 50 倍 |
| 规格化(PR) | 22 秒 | 0.3 秒 | 1 秒 | 73 倍 |
表 3:90000 细胞(消费级桌面:AMD 5950X + 64GB RAM + RTX 3090)
| 作用 | CPU | GPU | 加速 |
| 整个笔记本(不包括解耦器功能) | 774 秒(13 分钟) | 48 秒 | 16 倍 |
| 预处理 | 114 秒 | 6 秒 | 19 倍 |
| Regress out | 62 秒 | 1.6 秒 | 39 倍 |
| 主成分分析 | 42 秒 | 0.7 秒 | 60 倍 |
| HVG (Seurat v3) | 2.7 秒 | 0.4 秒 | 6.7 倍 |
| PCA | 175 秒 | 21.7 秒 | 8 倍 |
| Neighbors | 14.9 秒 | 4.6 秒 | 3.2 倍 |
| UMAP | 31 秒 | 0.3 秒 | 103 倍 |
| TSNE | 95 秒 | 1.4 秒 | 68 倍 |
| Louvain | 9.3 秒 | 0.5 秒 | 18 倍 |
| Leiden | 13.2 秒 | 0.1 秒 | 130 倍 |
| 逻辑回归 | 76 秒 | 3.75 秒 | 20 倍 |
| 绘图(FA2) | 191 秒 | 0.23 秒 | 830 倍 |
| run_mlm (DoRothEA) | 55 秒 | 12 秒 | 4.5 倍 |
| Run_wsum (程序) | 690 秒(11.5 分钟) | 28 秒 | 26 倍 |
9. 安装指南
安装 RAPIDS-singlecell 有多种方式:
- 使用 Conda yaml 文件(推荐):从 GitHub 仓库下载
conda/rsc_rapids_23.02.yml,然后运行:
conda create -f conda/rsc_rapids_23.02.yml - 从 PyPI 安装(需要预先安装 RAPIDS 和 CuPy):
pip install rapids-singlecell - 一次性安装所有依赖(包括 RAPIDS,实验性):
pip install 'rapids-singlecell[rapids]' --extra-index-url=https://pypi.nvidia.com
小提示: 如果采用第三种方法,请确保 CUDA 环境正确配置,以便 RAPIDS 和 CuPy 能够检测到 GPU。
10. 常见问题(FAQ)
- Q: cunData 与 AnnData 有什么区别?我应该在什么时候使用?
A: cunData 是 AnnData 的 GPU 轻量版,主要将数据存储在 GPU 内存中,特别适合需要大量矩阵运算的预处理和降维步骤。建议在预处理阶段使用 cunData,分析完成后通过.to_AnnData()转回 AnnData,以便利用 Scanpy 丰富的可视化功能。 - Q: 我的数据集有 50 万个细胞,消费级显卡(如 RTX 3090)能处理吗?
A: 可以。RAPIDS Memory Manager (RMM) 结合统一内存技术,允许 GPU 内存超额订阅并溢出到主内存。基准测试显示,RTX 3090 上 90K 细胞只需 48 秒,500K 细胞也能在 5 分钟内完成。 - Q: 我能否在已有 Scanpy 代码的基础上无缝迁移到 RAPIDS-singlecell?
A: 大部分情况可以。RAPIDS-singlecell 的 API 设计尽量贴近 Scanpy,例如rsc.pp.pca与sc.pp.pca参数相似。只需将 AnnData 转换为 cunData,然后调用 rsc 的函数即可。但需注意 cunData 的切片返回完整副本,而非视图。 - Q: peason_residuals 标准化在 GPU 上如何?
A: 通过normalize_pearson_residuals和highly_variable_genes的pearson_residuals口味,已实现 GPU 加速,在 500K 数据集上加速可达 73 倍。 - Q: 我是否需要安装 CUDA 和 cuDNN?
A: 是的,RAPIDS 依赖 CUDA 工具包和驱动程序。建议遵循 RAPIDS 官方安装文档,选择与你的 CUDA 版本匹配的包。
11. 结论
借助 RAPIDS-singlecell 库,你可以在比 CPU 仅计算 UMAP 嵌入所需时间更短的时间内,完成 500K 细胞的完整分析。这使得单细胞数据分析的迭代过程更加高效,生物信息学家能够实时与医生或生物学家协作分析数据。即使是消费级 3090 系列显卡,也能流畅分析 200K 细胞;而使用数据中心级 NVIDIA A100 80 GB GPU,你可以分析包含多达 2³¹-1(约 21.5 亿)个非零计数 的矩阵,轻松处理超过 100 万个细胞的数据集。
RAPIDS-singlecell 提供的 20 倍以上加速,让你能专注于分析和解释单细胞数据,而不是等待漫长的计算。这最终将显著提升生产力,并促进对细胞生物学的新发现。
你是一名 AI 行业编辑,请围绕下面这条热点输出一份资讯解读:
热点:RAPIDS实现单细胞数据分析速度提升的实用技巧要求:
1. 先用一句话解释这条热点在讲什么
2. 再总结它为什么重要
3. 说明会影响哪些 AI 产品或内容方向
4. 最后给出 3 个适合资讯站使用的标题
游乐网为非赢利性网站,所展示的游戏/软件/文章内容均来自于互联网或第三方用户上传分享,版权归原作者所有,本站不承担相应法律责任。如您发现有涉嫌抄袭侵权的内容,请联系youleyoucom@outlook.com。
 相关热点
相关热点佑驾创新与乐动机器人达成战略合作,围绕技术、产品、场景、数据四维度展开深度协同,旨在加速物理AI规模化落地,拓展无人车与机器人场景边界,推动具身智能商业化进程。
Meta开放AI算力租赁业务,市场反应从算力过剩转向算力商业运营。GPU从自用转向对外出租,算力从成本中心转为利润中心。AI云竞争核心从拥有GPU数量转向稳定跑满GPU的能力,依赖同步与参考时钟等底层基础设施的长期稳定运行。
针对大型多仓库工程(30+微服务、10+前端微应用),搭建包含规则、技能、子代理、13阶段工作流与门禁脚本的Harness系统,解决PRD不可信、方案与代码脱节、改完无人验证、交付环节琐碎等痛点,使AI在真实业务中稳定跑完需求。
部署MCP Toolbox前,先看清它的适用场景与安全边界,避免在权限管理不完善时接入敏感数据。 核心内容: 1 MCP Toolbox的核心功能与两种使用路线 2 项目适合与不适合的团队场景分析 3 实际验证的安全检查与关键限制 先说结论 MCP Toolbox 很适合小团队研究“让 AI
- 日榜
- 周榜
- 月榜
热点快看