




Nat Methods 人工智能如何精准预测大分子构象集合

蛋白质等大分子由多个动态构象组成,而当前主流方法主要捕获静态结构,难以完整描绘其动态变化与概率分布。预测完整构象集合面临定义不统一、实验数据整合困难及缺乏标准评估指标等挑战。未来需融合多源实验数据与计算方法,结合机器学习与分子动力学模拟,推动结构生物学从。
生命活动本质上是动态的。作为生命机器的核心执行者,蛋白质并非我们传统认知中固定不变的刚性结构。它们更像是一团持续流动、形态多变的“构象云”,由无数可以相互转化的微观状态共同构成。这个动态的、完整的构象集合,才是酶催化、信号转导、分子识别等关键生物学功能得以实现的真实物理基础。
然而,当前的结构生物学研究和以AlphaFold为代表的AI蛋白质结构预测,在很大程度上仍受限于“静态结构”的思维范式。AlphaFold的突破性成就,极大地提升了我们获取蛋白质高精度“静态快照”的能力,但它预测的本质上是一个最稳定的、时间平均化的构象。它无法揭示蛋白质在毫秒、微秒乃至更短时间尺度内如何运动,也无法告诉我们不同功能构象出现的相对概率。这正是下一代结构生物学与AI预测模型需要攻克的核心高地:精准预测蛋白质完整的、动态的构象集合。

长期以来,X射线晶体学和冷冻电镜(Cryo-EM)等技术为我们提供了海量高分辨率的蛋白质“标准结构”。这些静态结构极其珍贵,但它们通常只捕获了最稳定、最占主导地位的单一状态。在真实的细胞环境中,蛋白质无时无刻不在进行着多尺度的运动——从原子级别的微小振动,到二级结构单元的局部调整,再到结构域之间的大尺度开合与旋转。从统计物理和热力学的视角看,我们观测到的任何宏观生物学性质,都是背后无数微观构象状态按照玻尔兹曼分布进行加权平均的结果。
问题的关键在于,现有的实验技术各有其局限性,都只能窥见这个复杂动态景观的某一个侧面:
- 冷冻电镜和X射线晶体学能提供原子级分辨率细节,但样品处于冻结或结晶状态,大量动态信息因此丢失;
- 核磁共振(NMR)能够探测溶液中的动态行为,但其应用受限于分子量大小和信号灵敏度;
- 全原子分子动力学模拟理论上可以描绘构象变化的完整轨迹,却又受制于力场精度和有限的模拟时间尺度。
因此,我们目前掌握的多数“蛋白质结构”,很可能只是整个构象自由能图谱中少数几个能量最低的洼地。这种“静态结构中心主义”的视角,不仅限制了对蛋白质功能机制(通常依赖于构象间的切换)的深入理解,也框定了AI结构预测技术向更高阶发展的天花板。
蛋白质构象集合预测面临的核心挑战
那么,要构建一个能够预测构象集合的“AlphaFold 2.0”或下一代AI模型,我们需要跨越哪些根本性障碍?
首先,对于“构象集合”这一概念本身,不同领域的研究者尚未达成统一共识。部分研究仅关注少数几个宏观的、功能明确的状态,而忽略了其间大量连续变化的微观过渡态。然而热力学原理表明,即便是出现概率极低的构象,也可能对蛋白质的整体性质或功能通路产生不可忽视的影响,绝不能简单地只聚焦于“主角”构象。
其次,没有任何一种单一的实验技术能够独立、完整地捕获构象集合的全貌。每一种技术都像一盏特殊光谱的灯,只能照亮动态景观的特定部分。我们必须发展强大的数据整合能力,学会拼合这些来自不同光源的、互补但可能碎片化的信息。
第三,实验数据本身带来了巨大的解析挑战。绝大多数实验测量信号,都是时间上和体系内无数个分子状态的“ ensemble average ”(系综平均)。这导致了一个根本性的逆问题:可能有无数种不同的构象概率分布,都能产生完全相同的实验观测数据。从平均信号反推原始的构象分布,是一个典型的“病态逆问题”。
第四,我们严重缺乏统一的“描述语言”和“度量标尺”。现有的PDB数据库格式是为存储静态结构而设计的,无法有效表达复杂的、带有概率权重的构象集合。同时,我们也缺乏公认的、可靠的指标来量化比较两个构象集合之间的相似性。
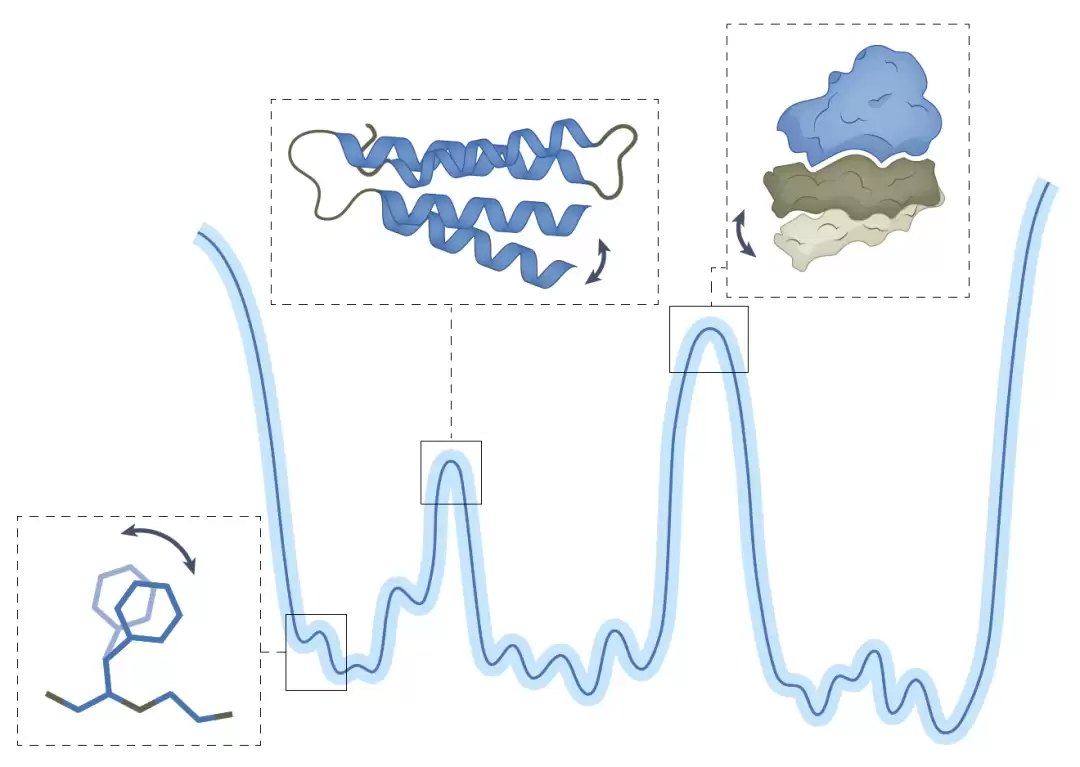
图1:蛋白质的自由能景观与其多尺度构象集合示意图。
如何构建可靠的构象集合“Ground-Truth”数据集
当务之急,是建立一套真正可靠、能被领域广泛认可的构象集合标准数据集,作为算法开发、训练与验证的基石。这要求我们彻底转变研究范式:从依赖“单一技术给出单一结构”转向倡导“整合结构生物学”。
我们需要像完成高难度拼图一样,系统性地整合不同技术提供的互补信息:
- 冷冻电镜和X射线提供高分辨率的静态结构框架;
- 核磁共振揭示局部原子的运动与动力学参数;
- FRET(荧光共振能量转移)、SAXS(小角X射线散射)等技术描绘大尺度的形状变化与距离分布;
- 分子动力学模拟则负责填充构象之间的转变路径与能垒信息。
以核糖体为例,其进行蛋白质翻译的效率和保真度,正是由多个构象状态(如A位、P位、E位的占据状态)之间的微妙平衡所决定,而这些状态又受到tRNA、mRNA及核糖体蛋白上微小涨落的显著影响。仅研究几个主要的“定格画面”,会错过大量调控翻译速率与准确性的精细分子机制。
事实上,许多现有的实验数据中,就隐藏着未被充分挖掘的动态信息宝藏。例如,冷冻电镜单颗粒分析数据中的颗粒异质性、X射线衍射数据中的漫散射信号、多温度晶体学中揭示的低概率构象状态等。未来,我们需要发展更强大的统计结构生物学算法与AI工具,从这些海量数据中高效、准确地提取出真正的构象动态信息。
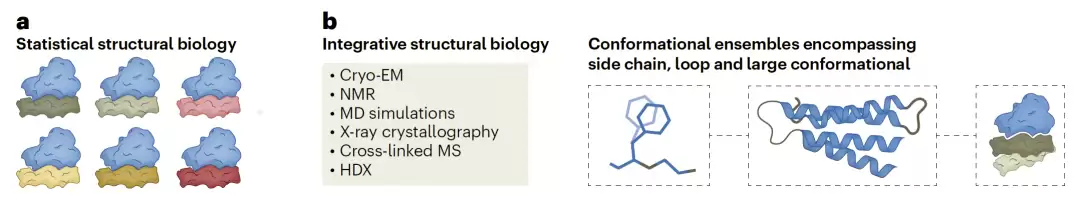
图2:统计结构生物学与整合结构生物学的研究框架。
实验数据的挑战:平均化、稀疏性与噪声
即便拥有了多源实验数据,重建构象集合依然困难重重。因为“系综平均”是绝大多数实验技术固有的特性。这意味着,截然不同的构象概率分布可能产生一模一样的实验信号;无处不在的实验噪声和误差会让问题变得更加棘手;而某些虽然总体占比较低、但对特定实验信号贡献极强的构象(例如在NMR的NOE实验中,由于信号强度与距离的六次方成反比,近距离接触的构象信号会被极度放大),会不成比例地主导观测结果。
为了应对这些挑战,贝叶斯推断、最大熵原理等统计物理方法正被引入结构生物学。它们能够在实验数据的约束下,对可能的构象状态进行重新加权和概率分配,从而推导出一个在物理上更合理、与所有数据最相容的概率分布。
此外,蛋白质内部往往刚性区域与高度柔性区域(如无序区域、柔性环区)并存,这使得传统的全局比较指标(如RMSD,均方根偏差)常常失灵。一个剧烈晃动的长环区产生的大幅度RMSD变化,可能完全掩盖了功能活性位点那些微小但关键的结构变化。因此,我们亟需发展一种层级化的构象表示与比较方法,能够同时处理不同空间尺度和时间尺度的运动,并将它们统一在同一个热力学分布框架之下。
构象集合的数据编码与相似性比较
目前,使用传统的PDB格式来表示复杂的构象集合,可以说是“小马拉大车”,力不从心。无论是用于表示替代构象的“altloc”字段、混合了动态信息与静态无序信息的B因子(温度因子),还是缺乏明确概率权重的多模型(multi-model)记录,都存在明显的缺陷和表达能力的局限。
未来的构象集合编码框架,必须能够满足以下核心需求:清晰表达构象状态之间的层级关系(如主链运动与侧链运动);明确区分构象本身的动态异质性和样品中不同组分的静态异质性;并且必须包含每个构象状态的玻尔兹曼权重及其不确定性的定量信息。
在相似性比较指标上,基于单一结构的RMSD显然已不适用。更合理的思路可能是基于概率密度函数的比较,例如计算两个构象分布之间的Jensen-Shannon散度或KL散度。但也有观点指出,仅依赖低维投影的概率密度函数可能会丢失关键的高维物理特征。或许,未来我们需要借鉴蛋白质语言模型的成功思路,发展出能够直接捕捉构象集合高维本质的嵌入表示方法,从而实现更智能的比较与检索。
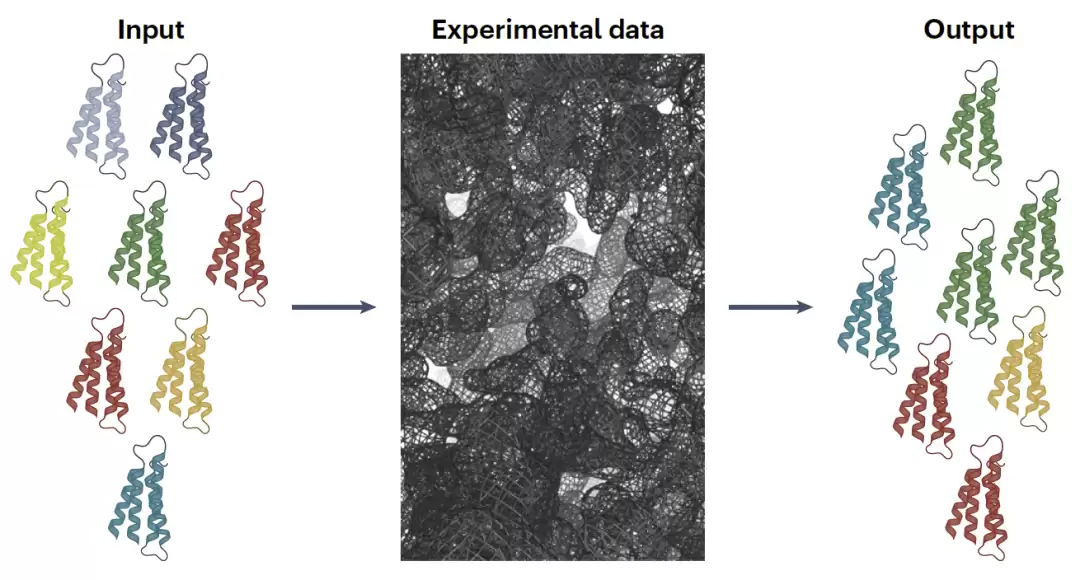
图3:从实验平均信号反演构象集合的病态逆问题示意图。
机器学习与分子动力学的深度融合之路
构象集合预测的终极解决方案,很可能在于机器学习(尤其是深度学习)与分子动力学模拟的深度融合。分子动力学模拟天生适合描述构象集合的采样与演化,但长期受限于采样效率和力场精度。如今,基于机器学习的力场正将模拟精度推向接近量子化学计算的水平,而增强采样方法(如元动力学)也借助深度学习实现了对稀有事件采样的突破。
更令人兴奋的是,生成式AI已经强势进军这一领域。例如,基于标准化流(Normalizing Flows)的Boltzmann生成器可以直接从平衡分布中采样生成构象;扩散模型(Diffusion Models)开始被用于生成具有多样性的蛋白质构象集合;一些前沿模型甚至已经能够预测内在无序蛋白(IDP)的构象系综分布。
不过,目前这些方法远未成熟。例如,通过人为操纵输入AlphaFold的多序列比对(MSA)信息,可以诱导它预测出不同的构象,但这些生成的构象缺乏真实的热力学概率背景,甚至可能产生能量上不稳定的非物理结构。因此,构建一个完善的数据、算法与实验闭环的基础设施至关重要——让AI辅助实验数据分析与解释,用高质量的实验集合数据优化和校正AI模型,再由新模型指导下一步的实验设计。未来的所有结构生物学数据,都应被视作“集合数据”来采集、分析和建模。
展望与讨论
我们正站在结构生物学一个历史性的转折点上:从“静态结构时代”迈向“动态构象时代”。研究的核心问题正在从“这个蛋白质的稳定结构是什么?”转变为“它如何在不同的功能状态之间动态切换?其构象能量景观的全貌是怎样的?”。
这一范式转变的影响将是深远而广泛的。精准的构象集合预测不仅能极大提升结构预测本身的精度与实用性,更可能彻底革新药物研发和蛋白质设计领域。例如,设计能够特异性稳定或扰动某个低概率构象(该构象可能与疾病状态相关)的变构药物;开发能够识别动态构象特征而非静态表位的新型抗体;以及从头设计能够穿越复杂构象能量景观以实现全新催化功能的人工酶。
目前困扰结构预测领域的许多难题,例如原子级别精度的进一步提升、RNA及其复合物结构预测的困境、以及点突变对蛋白稳定性和功能影响预测的失败,其深层根源可能都在于我们缺乏对目标分子完整构象集合的全面描述。因此,构建一个统一的大分子构象集合研究基础设施,涵盖从数据采集标准、整合建模方法、编码格式、比较指标、基准测试集到主动学习框架的全链条,已成为领域发展的当务之急。这场变革的最终目标,是让结构生物学真正成为一门研究“动态生命过程”的定量化、预测性科学。
参考资料
Wankowicz, S.A., Bonomi, M. From possibility to precision in macromolecular ensemble prediction. Nat Methods (2026).
https://doi.org/10.1038/s41592-026-03084-z
你是一名 AI 行业编辑,请围绕下面这条热点输出一份资讯解读:
热点:Nat Methods 人工智能如何精准预测大分子构象集合要求:
1. 先用一句话解释这条热点在讲什么
2. 再总结它为什么重要
3. 说明会影响哪些 AI 产品或内容方向
4. 最后给出 3 个适合资讯站使用的标题
游乐网为非赢利性网站,所展示的游戏/软件/文章内容均来自于互联网或第三方用户上传分享,版权归原作者所有,本站不承担相应法律责任。如您发现有涉嫌抄袭侵权的内容,请联系youleyoucom@outlook.com。
 相关热点
相关热点面壁智能聚焦端侧AI,不拼参数大小,而是通过知识密度提升与模型风洞技术,将大模型压缩至手机、汽车等设备。其MiniCPM以2B参数超越同期8B对手。CTO曾国洋22岁主导训练中国首个大语言模型CPM-1。端侧AI追求“默契系统”,在用户开口前预判需求,已在吉利、上汽大众等车型落地应用。
印度IT巨头HCLTech投资最高350亿卢比建设AI数据中心,容量可扩展至50MW,提供从设计到运营的端到端服务,旨在满足政府及企业日益增长的算力需求,抢占印度快速增长的数据中心市场,并推动AI基础设施布局。
小米具身机器人在汽车工厂自攻螺母上件工站实现双侧作业成功率98%,接近人工水平。同时在新工站分别达到90%成功率,从单一操作拓展至多工站协同,验证了具身智能在复杂工业环境的落地能力。
全球AI行业正迎来新的财富格局,DeepSeek创始人梁文锋凭借其公司的迅猛发展,个人财富急剧膨胀,一举超越多位硅谷知名人物,成为全球AI公司领域的新首富。以下将详细解析其身价飙升背后的关键因素及公司发展历程。 一、身价飙升至360亿美元,超越多位AI大佬 根据最新彭博亿万富豪指数,DeepSeek
- 日榜
- 周榜
- 月榜
热点快看