




多模态AI统一模型革新肽段质谱解析技术

pUniFind是多模态蛋白质组学基础模型,首次统一开放式肽段-谱图评分与零样本从头测序。基于超1亿条谱图训练,通过跨模态学习对齐谱图与序列信息。实验显示其在多个数据集中鉴定肽段数量超越传统方法,免疫肽组学提升42 6%,并能高效识别修饰与隐藏肽段,显著提升鉴定灵敏度与可信度。
蛋白质组学研究中,串联质谱(MS/MS)数据的精准解析始终是关键挑战。传统方法主要依赖数据库搜索和从头测序两大路径,但现有模型多局限于特征提取,缺乏一个能够统一打分、深度理解谱图与肽段关联的深度学习框架。这一瓶颈,如今被一项突破性研究——pUniFind模型所攻克。
近期,科研团队提出了大规模蛋白质组学基础模型pUniFind。其核心创新在于,首次实现了开放式端到端肽段-谱图匹配评分与开放式零样本从头测序的统一建模。简而言之,一个模型即可高效完成以往需要多个工具协同的两大核心任务。
该模型的强大源于其训练基础:它在超过1亿条经开放搜索标注的谱图海量数据上进行训练。通过精心设计的跨模态预测任务,pUniFind能够深度对齐质谱谱图与肽段序列这两种不同模态的信息,从而实现对肽段序列的精准预测和高效检索。
实际效果如何?数据最具说服力。凭借卓越的开放式评分能力,pUniFind在多个测试数据集上的肽段鉴定数量均超越了传统搜索引擎。尤其在极具挑战的免疫肽组学数据分析中,其鉴定数量提升了42.6%,增幅显著。
其从头测序能力更值得关注。团队设计了两种流程以适应不同场景。在修饰富集的从头测序任务中,即便搜索空间扩大300倍,pUniFind仍比现有方法多识别出60%的肽段-谱图匹配。在常规从头测序中,它额外恢复了38.5%的肽段,其中包含1891条能映射到基因组却不在参考蛋白数据库中的“隐藏”肽段,这对于新抗原发现等研究至关重要。
此外,团队开发的基于深度学习特征的质量控制模块,将测序结果与RNA-Seq证据的一致性从65.4%大幅提升至85.0%。可以说,pUniFind构建了一个统一且可扩展的深度学习框架,在鉴定灵敏度、修饰覆盖度和结果可信度方面均实现了跨越式提升。

要理解pUniFind的价值,需先回顾蛋白质组学数据分析的困境。串联质谱技术是现代蛋白质组学的基石,但传统解析工具如SEQUEST、MaxQuant、pFind等,其核心依赖于人工设计特征和相对简单的机器学习模型进行肽段-谱图匹配打分。
这种方法存在固有局限:通常假设固定的酶切规则,且仅支持有限的常见翻译后修饰。一旦遇到非特异性酶切或未知修饰,其性能便会显著下降。
为突破限制,学界发展了开放搜索技术(如Open-pFind、MSFragger)以更好地识别意外修饰和异常酶切。另一方面,完全无需数据库的从头测序技术在抗体测序、新抗原发现等领域价值独特,催生了DeepNovo、Casanovo等深度学习模型。然而,这些方法往往各自独立,准确率和鲁棒性仍有提升空间。
近年来,深度学习在谱图预测、保留时间预测等单任务上展现出潜力。学界逐渐认识到,数据库搜索和从头测序本质都依赖于对谱图与肽段关系的深度建模。但一直缺乏一个统一框架,能将搜索、测序及质量控制任务整合。与此同时,多任务与多模态学习在计算机视觉、蛋白质设计等领域已证明其强大的泛化能力。
于是,一个清晰的思路应运而生:能否构建一个统一的多模态深度学习模型,一举提升数据库搜索和从头测序能力,推动蛋白质组学解析从“特征工程”时代迈向“端到端学习”新阶段?pUniFind正是对这一问题的完美解答。
方法:统一的多模态预训练框架
pUniFind的架构设计充分体现了“统一”思想。模型核心是分别对肽段序列和MS/MS谱图进行编码,然后通过多个跨模态预训练任务促使它们深度对齐。这些任务包括从头测序、谱图预测及候选肽段排序,让模型从多角度学习谱图与肽段的关联。
为训练此模型,研究团队利用Open-pFind对大规模公开数据进行了重新注释,构建了包含超1亿条肽段-谱图匹配的训练集,庞大的数据规模奠定了其作为“基础模型”的基石。
在模型结构上,谱图编码器需学习谱图的整体特征,并精细理解每个峰可能对应的氨基酸数量、离子类型及肽段长度。肽段编码器则负责生成理论谱图表征。随后,一个联合模态评分器会融合两者的嵌入信息,实现端到端的匹配评分。团队还引入了列表排序策略,增强了模型对候选肽段间相对关系的建模能力,使打分更为精准。
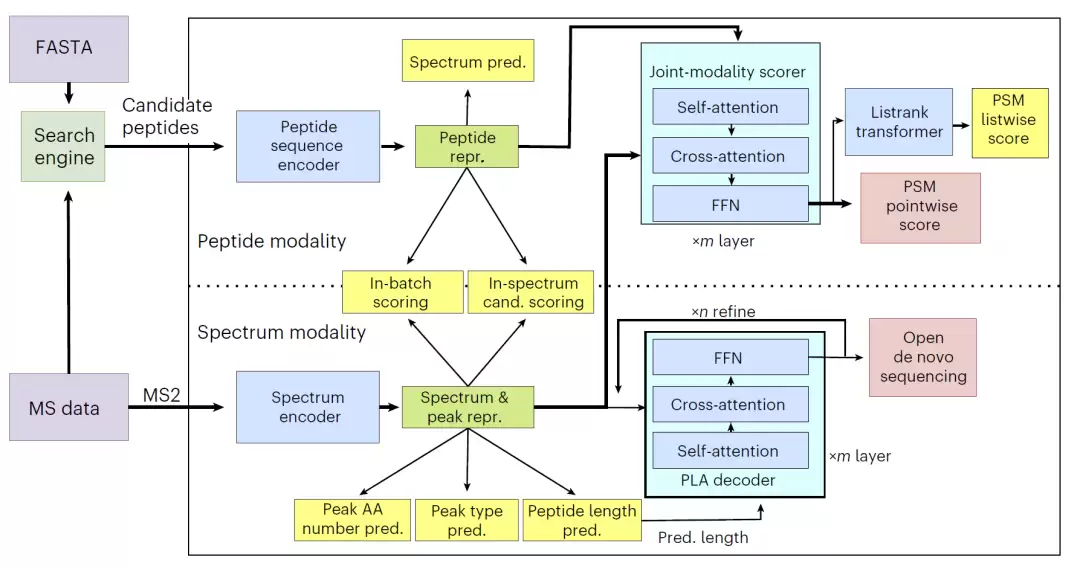
图1:pUniFind 模型整体架构与多任务训练流程。
结果:全面超越的性能表现
在多物种数据集上实现更高肽段鉴定率
研究首先在九个不同物种的数据集上检验了pUniFind的数据库搜索能力。结果显示:在开放搜索模式下,pUniFind在所有数据集上均获得了最高的肽段鉴定数量,相比优秀工具Open-pFind,提升幅度在2%到18%之间。尤其在Vigna mungo和Bacillus subtilis数据中,提升最为显著。
进一步的消融实验揭示了性能提升的来源。当引入从头测序和谱图预测这两个关键跨模态训练任务后,模型在枯草芽孢杆菌数据集上的肽段鉴定能力提升了60%。这有力证明,跨模态学习切实增强了模型对谱图-肽段关联的本质理解。
为评估模型可靠性,团队采用了“诱饵数据库”策略。在混入蜜蜂蛋白作为干扰的实验中,pUniFind即便鉴定出更多肽段,其错误匹配比例仍保持正常甚至更低,表明其高灵敏度并未牺牲准确性,鲁棒性值得信赖。
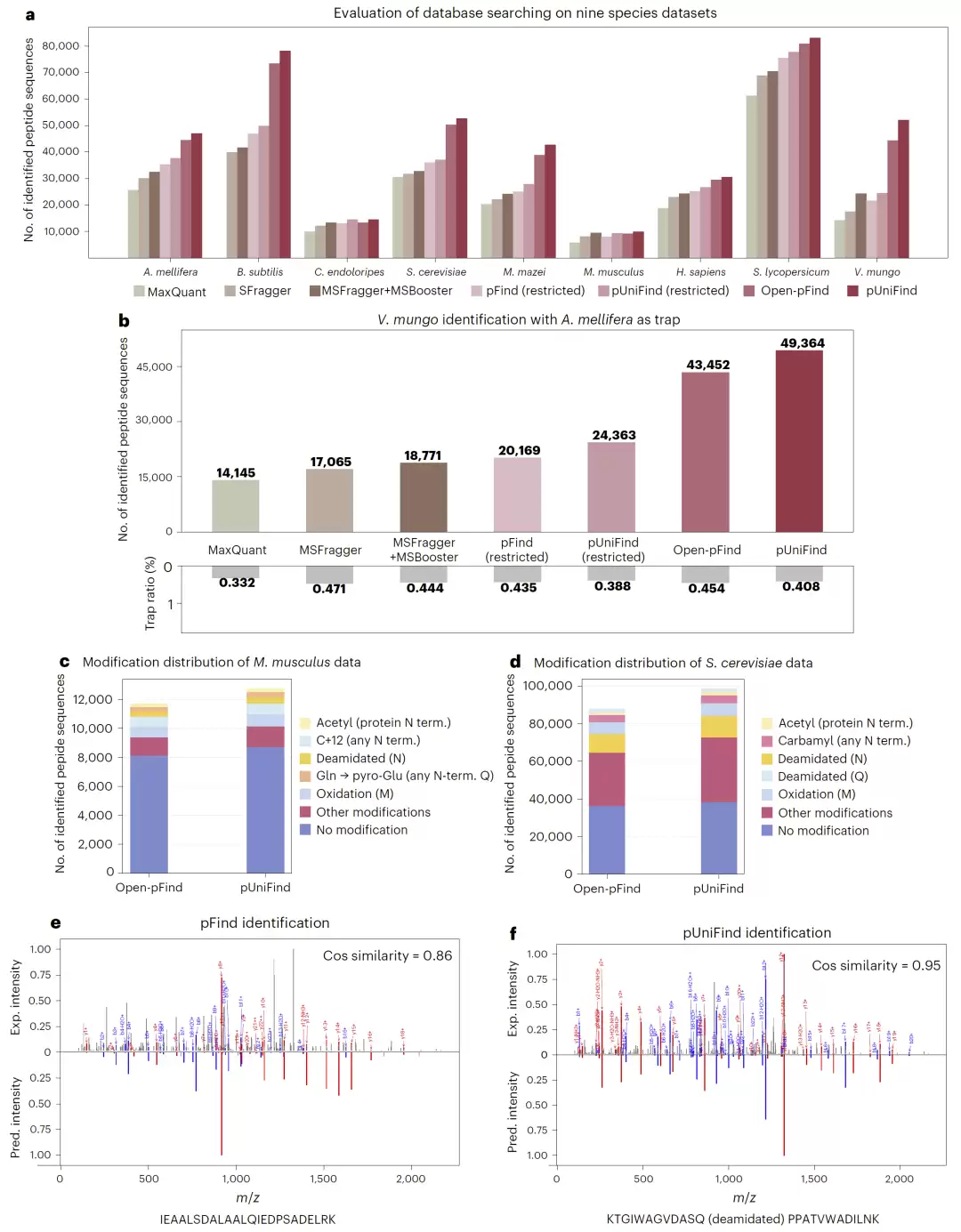
图2:pUniFind 在九个物种数据集中的数据库搜索表现与修饰肽段分析。
深度学习跨模态训练显著提升修饰肽段识别能力
pUniFind在处理翻译后修饰方面展现出独特优势。在修饰肽段占比高达58.4%的酵母数据集中,其性能提升更为明显。与Open-pFind基于SVM的评分方式不同,pUniFind通过谱图预测任务,能够避免对低频修饰产生不合理惩罚,从而更稳定地识别罕见修饰类型。
在一个包含21种不同修饰的数据集测试中,即使不进行在线优化,pUniFind仍在61.9%的修饰类别中获得了更高的肽段识别数量,显示出对多样化修饰类型的广泛适应能力。
在新型仪器数据上维持高精度
研究也考察了模型在Astral和timsTOF等新型质谱仪数据上的表现。经微调后,pUniFind在Astral数据上的性能相比Open-pFind和MSFragger+MSBooster组合提升了约9%。
在利用代谢标记技术的大肠杆菌数据集中,pUniFind鉴定出的肽段-谱图匹配数量比pFind高10%,比MSFragger+MSBooster高43.9%,同时其缺失定量比例低至0.26%。这表明它不仅在“量”上取胜,在“质”上也保持了高水准。额外的混合物种诱饵测试进一步证实,pUniFind未因训练数据产生明显的标签记忆,其评分策略更为保守可靠。
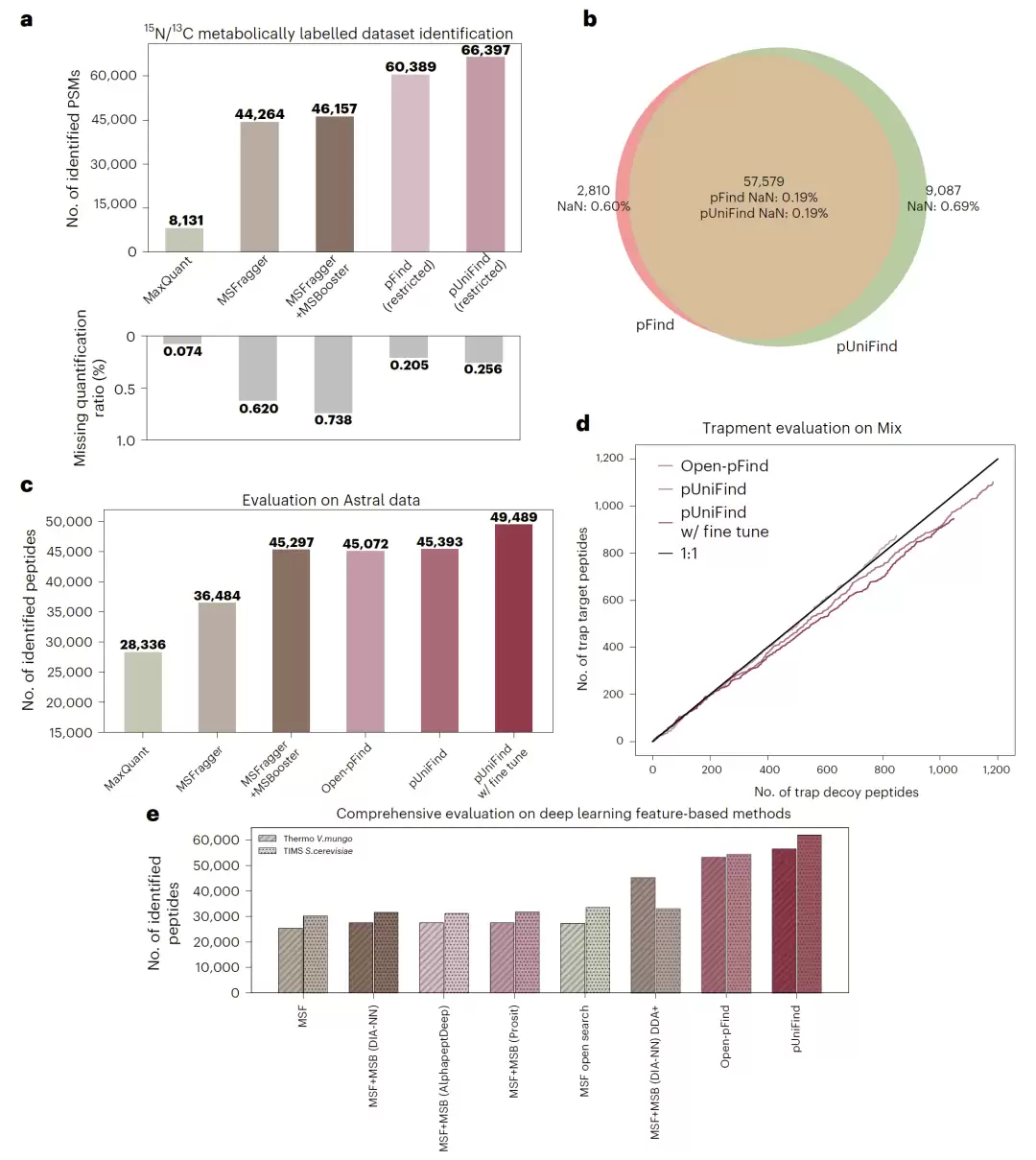
图3:pUniFind 在 Astral、TIMS 与代谢标记数据中的性能评估。
构建开放式从头测序统一工作流
针对从头测序,团队提出了两种模式:常规模式适用于一般数据集,而富含修饰的模式则针对磷酸化等修饰富集的数据进行了优化。为提升结果可信度,他们还开发了一套基于深度学习特征的过滤策略,综合端到端评分、谱图预测和保留时间等多维度信息,系统性地过滤低可信结果。
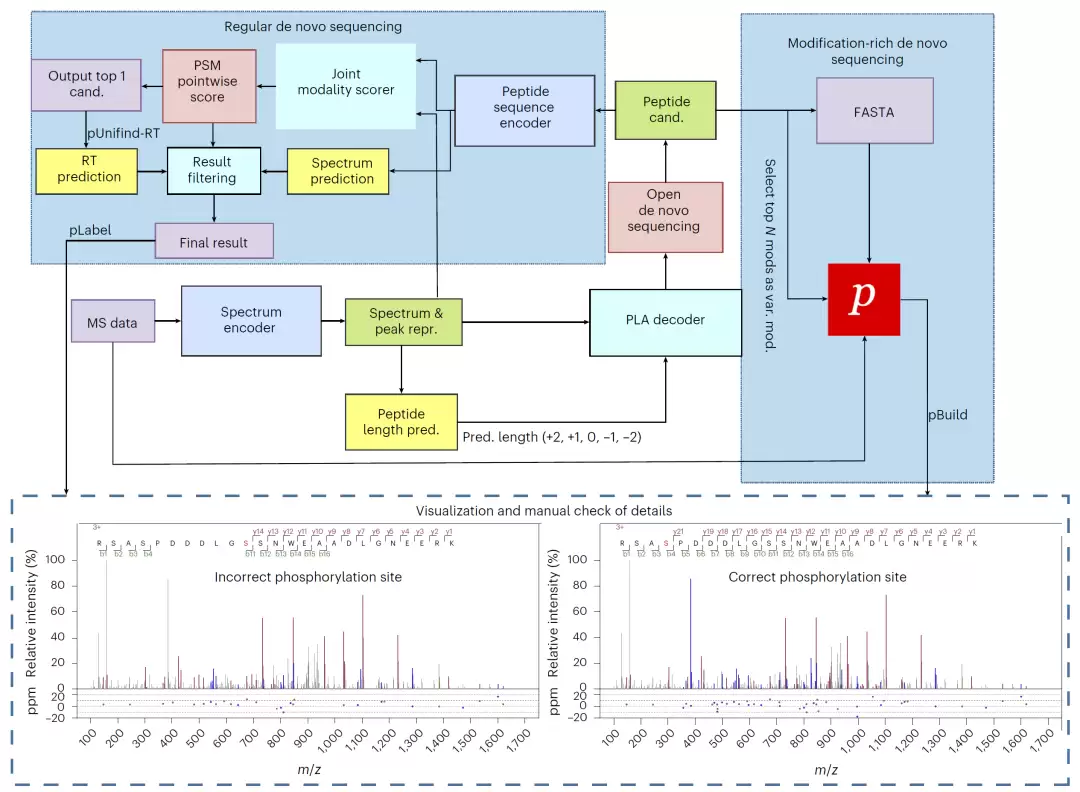
图4:pUniFind 开放式 de novo 测序工作流。
在富修饰数据集上显著优于传统方法
在包含21种修饰的基准测试中,pUniFind+pFind组合的平均肽段召回率达到63.8%。即便搜索空间扩大300倍,其性能仍与传统从头测序方法持平,并比pNovo高出60%。
一个关键优势是,pUniFind可以在没有任何先验知识的情况下,同时考虑超过1300种潜在修饰。作为对比,pNovo即使提前告知正确的修饰类型,其召回率也只有47.6%。在修饰水平、序列水平和位点水平三种评估标准下,pUniFind均明显胜出。
更令人印象深刻的是,即使是训练集中样本极少的稀有修饰,pUniFind依然能获得超过60%的肽段召回率,这说明模型对低频事件具备了出色的泛化能力。
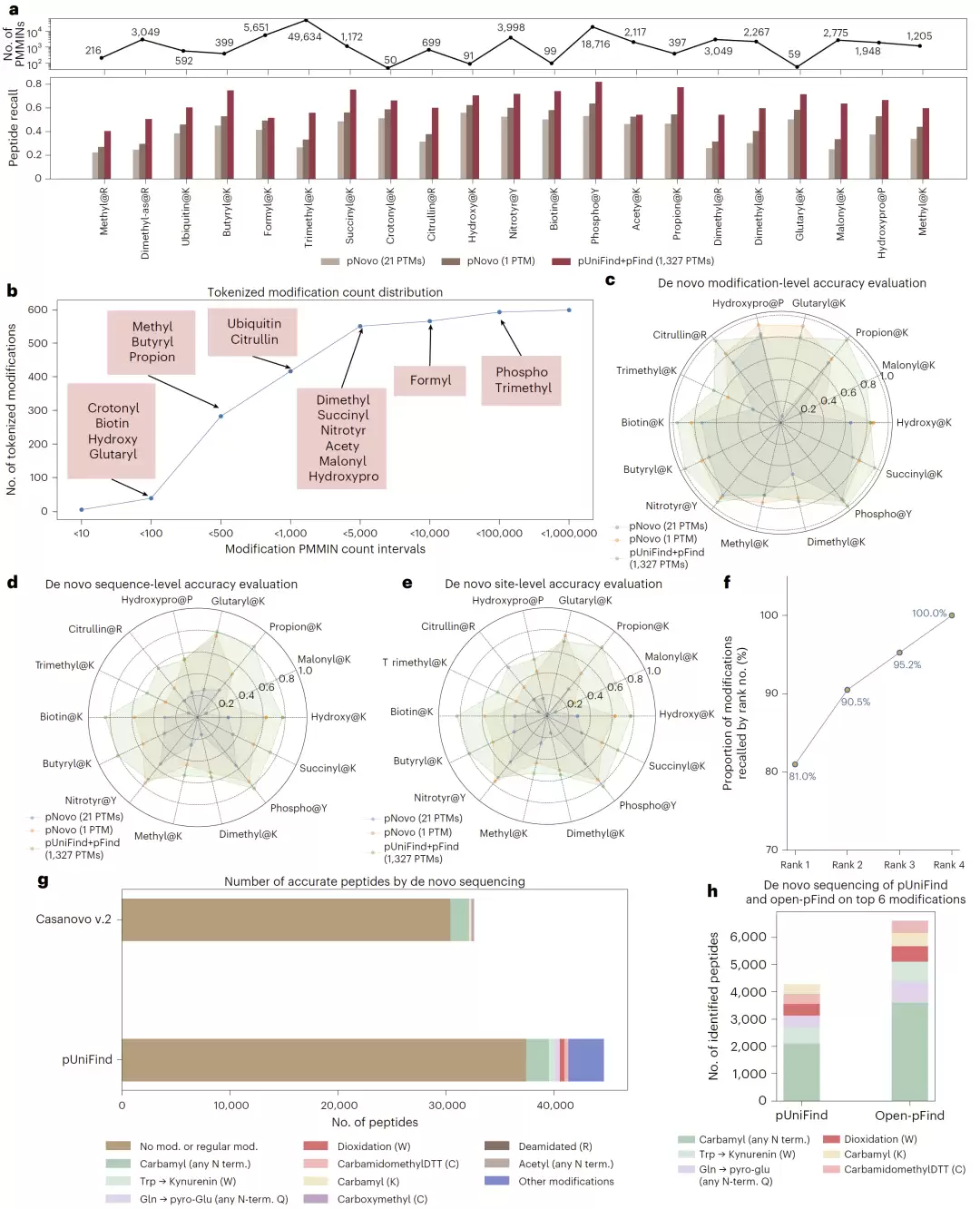
图5:pUniFind 在21种 PTM 数据集上的开放式 de novo 测序性能。
在复杂应用场景中取得突破
研究进一步将pUniFind推向更具挑战性的应用场景,如宏蛋白组学和免疫肽组学。在宏蛋白组学数据中,pUniFind比Open-pFind和MSFragger+MSBooster分别多识别出6.3%和29.0%的肽段。
在难度更高的免疫肽组学数据中,其鉴定数量相比MSFragger+MSBooster提高了17.4%,相比Open-pFind更是提高了42.6%。同时,这些新增鉴定谱图的余弦相似度中位数高达0.95,证明新增结果具有很高的可靠性。
在从头测序任务中,pUniFind的肽段召回率达到86.3%,显著高于Casanovo v2的69.1%。研究还发现,模型能识别出大量不存在于参考蛋白数据库、却能映射到人类基因组的HLA肽段,其中1891条满足高可信标准,为新抗原发现提供了宝贵资源。
此外,基于RNA-Seq支持数据集的评估表明,其过滤模块可将结果与RNA-Seq证据的一致性从65.4%大幅提升至85%,极大增强了从头测序结果的可信度。
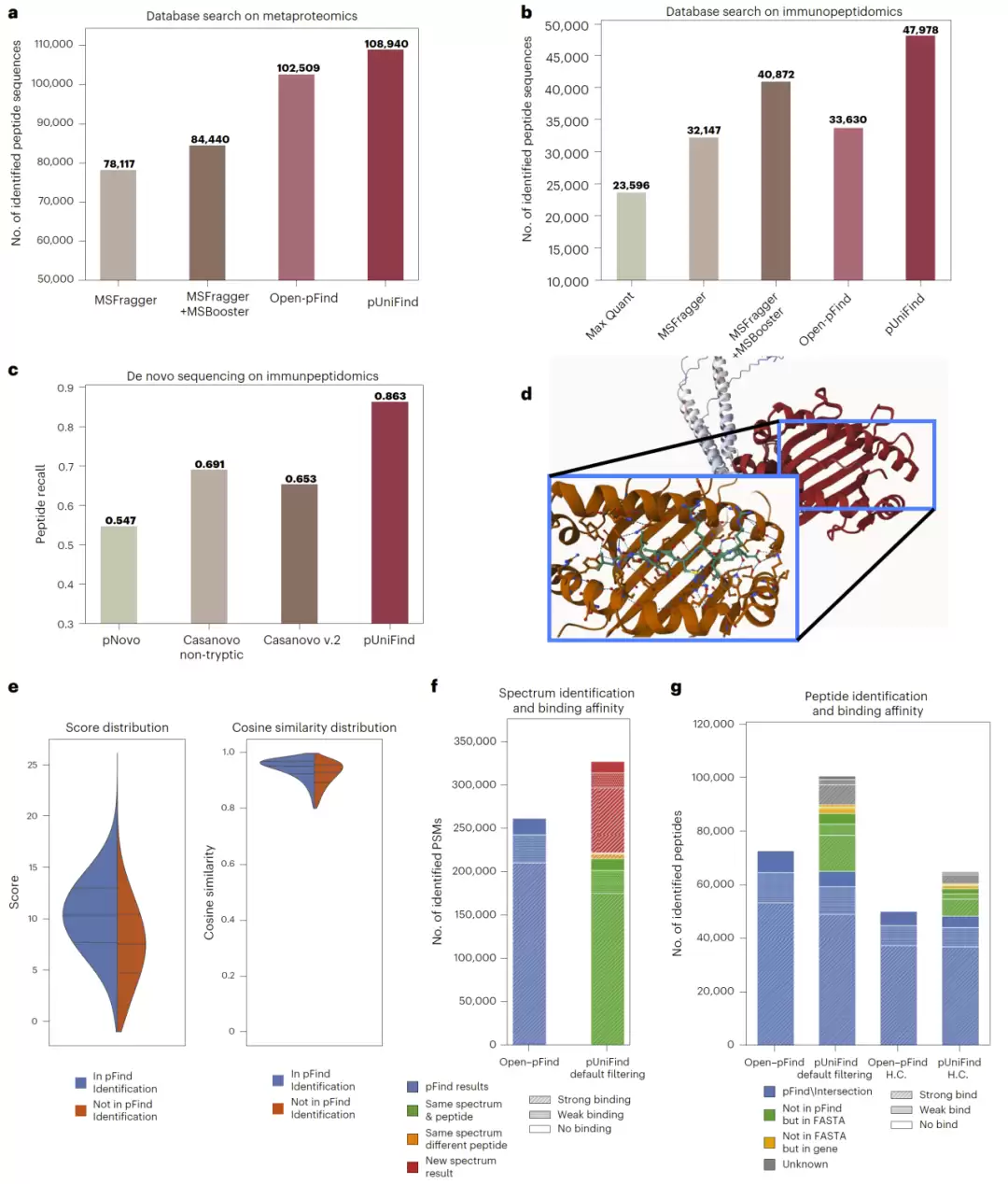
图6:pUniFind 在免疫肽组学与宏蛋白组学中的应用表现。
讨论与展望
pUniFind研究首次将开放数据库搜索、开放从头测序以及深度学习质量控制统一到一个多模态预训练框架中。通过跨模态预训练,模型实现了对谱图和肽段信息的同步深度理解,完成了真正意义上的端到端肽段-谱图匹配评分。结果表明,这种统一的深度学习框架不仅优于传统的特征工程方法,更展现出替代当前主流评分体系的潜力。
特别是在免疫肽组学、宏蛋白组学等搜索空间巨大、背景复杂的场景中,pUniFind的优势得到了充分体现。此外,它实现的开放式从头测序,使得修饰肽段的测序性能首次达到甚至超过了未修饰肽段的水平,这是一个重要的里程碑。
当然,研究团队也指出了当前模型的局限。例如,在数据库搜索任务中,保留时间信息尚未被充分整合;模型也尚未完全适配数据非依赖采集模式的分析。未来的工作将向大规模DIA数据集拓展,并进一步完善这个统一的谱图解析框架。可以预见,随着数据规模和模型能力的持续增长,蛋白质组学数据分析的自动化与智能化程度将迈向新的高度。
参考资料
Zhao, J., Mao, P., Wang, K. et al. A large-scale unified deep learning model for peptide mass spectrum interpretation trained on multimodal data. Nat Mach Intell (2026).
https://doi.org/10.1038/s42256-026-01234-8
你是一名 AI 行业编辑,请围绕下面这条热点输出一份资讯解读:
热点:多模态AI统一模型革新肽段质谱解析技术要求:
1. 先用一句话解释这条热点在讲什么
2. 再总结它为什么重要
3. 说明会影响哪些 AI 产品或内容方向
4. 最后给出 3 个适合资讯站使用的标题
游乐网为非赢利性网站,所展示的游戏/软件/文章内容均来自于互联网或第三方用户上传分享,版权归原作者所有,本站不承担相应法律责任。如您发现有涉嫌抄袭侵权的内容,请联系youleyoucom@outlook.com。
 相关热点
相关热点面壁智能聚焦端侧AI,不拼参数大小,而是通过知识密度提升与模型风洞技术,将大模型压缩至手机、汽车等设备。其MiniCPM以2B参数超越同期8B对手。CTO曾国洋22岁主导训练中国首个大语言模型CPM-1。端侧AI追求“默契系统”,在用户开口前预判需求,已在吉利、上汽大众等车型落地应用。
印度IT巨头HCLTech投资最高350亿卢比建设AI数据中心,容量可扩展至50MW,提供从设计到运营的端到端服务,旨在满足政府及企业日益增长的算力需求,抢占印度快速增长的数据中心市场,并推动AI基础设施布局。
小米具身机器人在汽车工厂自攻螺母上件工站实现双侧作业成功率98%,接近人工水平。同时在新工站分别达到90%成功率,从单一操作拓展至多工站协同,验证了具身智能在复杂工业环境的落地能力。
全球AI行业正迎来新的财富格局,DeepSeek创始人梁文锋凭借其公司的迅猛发展,个人财富急剧膨胀,一举超越多位硅谷知名人物,成为全球AI公司领域的新首富。以下将详细解析其身价飙升背后的关键因素及公司发展历程。 一、身价飙升至360亿美元,超越多位AI大佬 根据最新彭博亿万富豪指数,DeepSeek
- 日榜
- 周榜
- 月榜
热点快看